Formal metadata describing these data are available.
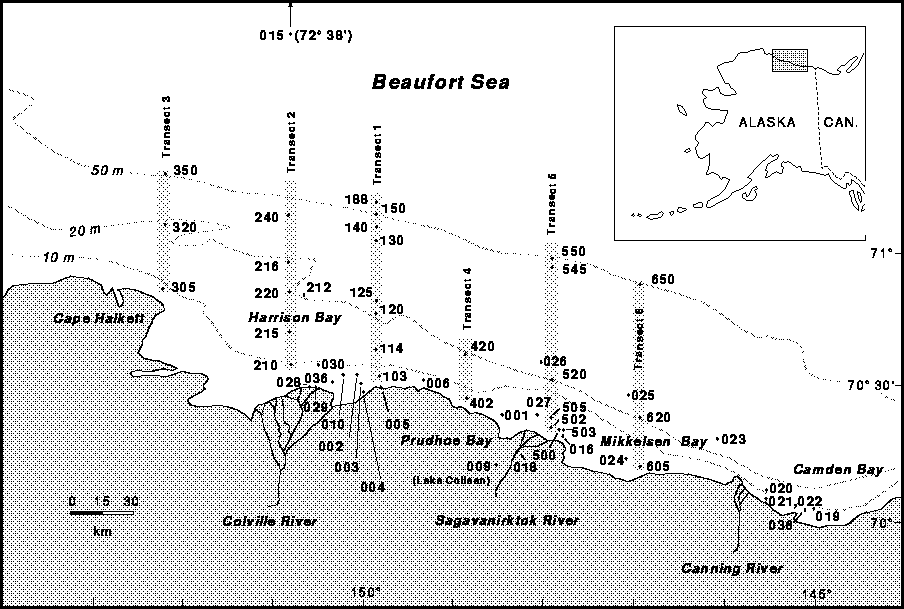
Gas hydrate deposits of polar continental shelves are currently believed to be the most vulnerable to climate change (Kvenvolden, 1988). These areally extensive shelves, formerly exposed to very cold surface temperatures (yearly average -10° to -20°C) have been and are being transgressed by a much warmer polar ocean (~0°C). As a result, the polar shelf surface in water depths greater than 5 m, has experienced an approximately 10°C or more temperature increase over the past 10,000 years. Although pressure on shelfal gas hydrate deposits has increased owing to a rise in sea level of about 100 m, this pressure increase which would stabilize the gas hydrate, is more than offset by the large temperature increase that destabilizes the gas hydrate. The amount of methane released by this process has been estimated to be about 4 Tg (1012 grams) year-1 (Kvenvolden, 1991) or about 1% of all current sources of atmospheric methane. If this suggestion and estimate are correct, then escape of methane from gas hydrate deposits of polar continental shelves should be observable. To test this idea, beginning in 1990 we conducted surveys of methane in sediment, water, and ice in an area of the Beaufort Sea continental shelf offshore from the north coast of Alaska. Previously, measurements of atmospheric methane concentrations at Barrow, Alaska, about 200 km NE of the study area, showed a seasonal increase during August through October, perhaps due in part to methane venting from Arctic shelfal waters (figure 1). This report summarizes our data collected from 1990 through 1994.
These data are from Dlugokencky et al. (1994) and can be obtained through anonymous ftp from cdiac.esd.ornl.gov.

Methods
In our survey, we measured methane concentration in sea water under the
ice on six primary transects from nearshore to the shelf break at water
depths ranging from 2.5 to 88m from Cape Halkett east to Mikkelsen Bay,
Alaska (figure 2). Other surface water samples were taken at various
locations where water depths ranged from 2 to 20m within the area between
Camden Bay and the Colville River Delta (figure 2). One oceanic surface
water sample was also collected in April 1993 at 72° 38' N. latitude .
The surveys were undertaken during the last week of April and first week
of May 1992, the middle of April 1993, the latter portion of April 1994,
the second week of September 1993, and the later portion of August 1994.
At each station in April and May, a 23-cm diameter hole was drilled
through the 2-m thick first-year ice. Niskin bottles (1.7 l) were
attached at intervals to a line lowered to the seafloor and subsequently
tripped to collect water samples for methane determinations. In September
1993 and August 1994, when no ice was present, a boat was used to transit
to stations. As each Niskin bottle was recovered, 100 cm3 of water was
transferred to a 140 cm3 syringe. A heat lamp to prevent the water from
freezing during April and May surveys. In the laboratory at the end of
each day's field work, 40 cm3 of ultra-pure nitrogen were added to each of
the syringes, and methane was extracted from the water into the nitrogen
at room temperature by shaking the syringe, following a method modified
from McAullife (1971). A portion of the resulting gas mixture was
measured for methane content by gas chromatography. Multiple extractions
were used on several samples to determine an empirical extraction
efficiency coefficient which was utilized with the remainder of the
samples that were extracted only once. Our analyzed gas standard
contained 10 ppm ±2% methane in nitrogen. Duplicate samples were
analyzed for each water depth with an average standard deviation of
±0.6 nM.
CTD (conductivity, temperature, depth) casts were made at each station. CTD data was recorded internally by a Seacat 19 CTD probe made by Sea Bird of Seattle Washington. The data was downloaded onto a computer where it was processed using Seasoft Version 4.016. The sub-programs LOOPEDIT, BINAVE, then DERIVE were utilized to process the raw data. The data were averaged into 0.5m intervals. The upcast and downcast were evaluated, and only one cast is plotted. In general the upcast was used because the sensors showed less fluctuation. The results are plotted for each station and appended with water methane concentrations in Appendix 1.
Digital data from the CTD casts are available online by individual cast as tab-delimited ASCII (plain text) files or in their entirety as a UNIX-style archive (tar format).
The methods utilized to measure nutrients in this study were adapted from older colorimetric procedures and are described in detail by Whitledge and others, (1981). A basic description of each method, conducted on a Technicon Autoanalyzer System, follows.
Phosphate is determined as phosphomolybdic acid which in its reduced form in the presence of antimony has an absorption maximum at 880 nm. The method is basically an automated version of the procedure of Murphy and Riley (1962).
Orthosilicic acid is determined by its reaction with molybdate in aqueous acidic solution to form silicomolybdic acid. In this procedure, which is basically that of Armstrong and others, (1967), stannous chloride is used to reduce silicomolybdic acid to the heteropolic acid which has an absorption maximum at 820 nm.
Nitrite is determined by the Greiss reaction in which sulfanilamide and N-(1-Napthyl)ethylenediamine dihydrochloride react with nitrite in aqueous acidic solution to form an intensely pink diazo dye with an absorption maximum at 540 nm (Bendschneider and Robinson, 1952). Nitrate, after it is reduced to nitrite by passage through a column of copperized cadmium filings, is determined in an identical manner to nitrite (Wood and others 1967). This analysis gives the sum of nitrate + nitrite, and nitrate is determined by difference.
Ammonium is determined by the Berthelot reaction in which hypochlorous acid and phenol react with ammonium in aqueous alkaline solution to form indophenol blue, an intensely blue chromophore with an absorption maximum at 637 nm. The method utilized is a modification of the procedure reported by Slawyk and MacIsaac (1972).
The methane content of sea ice was measured at 51 stations in 1993 and 1994. A 1m long by 7.6 cm diameter ice corer with an extension was used to core through the ice to sea water. The ice core was laid out on the sea ice, measured and cut into 10 cm sections. Selected intervals were chosen, then placed in 1-liter friction sealed cans equipped with 2 septa ports, sealed, and kept frozen. At the field laboratory, each sample was weighed, then the headspace within the can was purged with ultra pure nitrogen for 5 minutes at flow rates exceeding 200 ml/min. Tests on the methane content of the purged headspace with the frozen ice core inside yielded concentrations less than 0.1 ppm. The cans containing ice samples were placed in hot water baths and stabilized at approximately 20° C. Each can was shaken by hand for about 30 seconds to partition the dissolved methane between the water and the nitrogen headspace. Next, a syringe containing 30 ml of ultra pure grade nitrogen was injected into the can, then 30 ml of the resulting mixture of headspace and gas was removed and analyzed for methane content in the same manner as described for sea water samples. The volume of headspace in the can was determined by the weight of the ice sample, assuming that once melted the water occupied a volume of 1 ml/gm.
Sediment samples were taken in May 1992 operating from the ice canopy. A
hole was augured in the ice, through which a 45 x 7.5 cm core barrel with
an auger cutting head was lowered. The core barrel was attached to 1.5-m
long connecting stems in series up to a maximum length of 15.5 m. Once on
the ocean bottom, the core barrel was rotated by hand with the aid of a
T-bar until no further penetration was achieved. Upon retrieval, the
liner containing sediment was extruded onto the ice. The least disturbed
10-cm long (a volume of about 450 ml wet sediment) section of the core was
chosen and placed in a septa-equipped 1-liter sample can. Seawater was
added to the brim of the can, then 200 ml of water was removed, creating a
200 ml headspace. About 2-3 grams of sodium azide was added as a
biocide. The can was sealed, then purged with helium at a flow rate
estimated to be greater then 300 ml/min. for 5 minutes. The samples were
kept frozen until analysis at our Menlo Park laboratories.
Results
Stations in water depths from 2.5 to 88m were established on six north-
south transect lines as shown in figure 2. Three digit, abbreviated
station numbers are shown on the transect lines. Many stations were
reoccupied on subsequent surveys. Each station is given 3 additional
numbers making a total of six digits per station. The first two refer to
the year, the third to the month of sample collection, the fourth to the
transect number, and the last two represent the approximate water depth at
each station. For example, station 925150 can be read as 1992, May,
Transect 1, at 50 meters water depth. When the same station was
reoccupied on a subsequent survey only the first 3 digits change denoting
a different sampling date; i.e., 934150 was completed in April, 1993.
Stations off transect lines are given a 0 in the fourth digit place.
Station locations are available in a table in tab-delimited ASCII, with the station locations encoded as decimal degrees north latitude and west longitude.
Water Methane Concentrations
Water methane concentrations and methane oxidation rates are listed in
Table 1, and are illustrated by graphs of methane vs. water depth at each
transect in Appendix 2. Measurements of methane in ice-covered sea water
were made in May 1992, April 1993, and April 1994. The results are
compared with measurements taken in ice-free water during September 1993
and August 1994.
Analyses of 143 samples from ice-covered waters demonstrate that methane concentrations in the water column are supersaturated with respect to the equilibrium solubility of methane with the atmosphere (~4 nM). Maximum methane concentrations at a given station ranged from 12 to 275 nM and were typically about 15 to 25 nM throughout the water column. The highest methane concentrations were found near the bottom. Maximum methane concentrations are given in map view for each survey in Appendix 3.
Methane concentrations from ice-free water were determined in 103 samples. Maximum methane concentrations at a given station ranged from 5 to 44 nM with one anomalous measurement of 148.6 nM at station 948120, 18m water depth. Anomalous methane concentrations (33.7-94.4 nM) were also observed at this same station, 925120 throughout the water column in May, 1992. Typically, methane concentrations were at a minimum at the surface, remained nearly constant or increased with depth to concentrations ranging from 9 to 56.4 nM, excluding the 148.6 nM anomaly mentioned above. Methane concentrations in the upper 3m of ice-free water never exceeded 18 nM and were commonly between 6 and 14 nM.
A limited number of measurements of the carbon isotopic composition of methane were made at the University of Hawaii. The results shown in Table 1 reveal a wide range of values from -31.1 to -80.5 o/oo.
Methane Oxidation rates were determined in selected samples (Table 1) and are given graphically in Appendix 4. The rates during ice covered times were higher than most measurements made worldwide. In ice free periods measured to date (September, 1993), the oxidation rates were undetectable. Thus a seasonality exists regarding the oxidation of methane by bacteria living in the water column.
Column headings:
Column headings:
We sampled fast ice and seasonal pack ice at 36 locations along the coast from Harrison Bay to Camden Bay in water depths ranging from 1.8 to 57 m. Maximum methane concentrations are given in Appendix 5 and the complete results are listed in Table 3. Methane concentration profiles along transects are given in Appendix 6.
Methane concentration profiles through the ice within the upper 50 cm interval and from water depths less than about 10m contained the highest concentrations of methane (7 to 1260 nM), with typical values of about 30 to 40 nM. In the ice interval from 50 to 200 cm, the methane concentration ranges from 5 to 286 nM, with typical values of about 15 nM.
Ice that formed in water depths greater than 15m shows the same trend of decreasing methane with increasing depth in the ice core; however, the methane concentrations were less varied and were always lower. Within the 0 to 50 cm interval methane concentrations ranged from 8 to 14 nM and from 5 to 14 nM within the 50 to 200 cm interval. Measurements of the carbon isotopic composition of methane in ice are shown in Table 3 and reveal a wide range of values from -52.1 to -83.4 o/oo.
Column headings:
Column headings:

Methane concentrations range from 2.2 to 10.1 ml/l wet sediment. There is no distinctive pattern of methane concentration. However the highest concentrations were recorded within the confines of one mapped shallow gas anomaly. The highest concentrations are found near the Colville River Delta and range from 4.6 to 10.1 ml/l. The lowest concentrations of methane (3.8 and 2.2 ml/l) were found offshore at Weller Bank in clean sand. This low concentration is perhaps the result of methane escaping more readily from the clean sand rather than silt or clay which comprise the bulk content of the rest of the samples.
Summary
This report documents the concentrations of methane in sediment, water,
and sea ice in an area of the Beaufort Sea continental shelf offshore
northern Alaska. Data were collected during field surveys conducted from
1990 to 1994. In addition to methane concentrations, information
regarding methane oxidation rates, nutrients, salinity, and temperature
of the water column is also tabulated. In general, more methane is
present in the water when ice forms a seal over the water column than when
ice is absent. Methane is also present in ice often in concentrations
exceeding those in water. Fast ice forming in place over water depths
less than about 10m can contain methane concentrations an order of
magnitude higher than that of the underlying water. Measurements of the
carbon isotopic composition of methane and ice were initiated in 1994 as
an attempt to define the source or sources of methane. These data
represent the first ever collected is such a setting. The data thus far
demonstrate a wide range of values from -31.6 to -83.4 o/oo indicating
that there a may be complex variety of sources.
Acknowledgments
The project has been conducted in collaboration with the University of
Washington and the University of Hawaii. We thank (1) M.D. Lilley, E.J.
Olson, and E. McLaughlin, School of Oceanography, University of
Washington, for the information on methane concentrations, methane
oxidation rates, and nutrient concentrations in seawater; (2) B.N. Popp,
F. J. Sansone, and T. Rust, SOEST, University of Hawaii, for the isotopic
analyses of methane. We are grateful to (1) P.W. Barnes, U.S. Geological
Survey, for guidence in Arctic operations, and (2) E. Reimnitz for help in
collecting sediment samples for gas analyses. This work is supported by
the USGS Global Change and Climate History Program.
Keith A. Kvenvolden
Mail Stop 999
U.S. Geological Survey
345 Middlefield Road
Menlo Park, CA 94025
Tel: (415) 354-3213
FAX: (415) 354-3191
email: kkvenvolden@usgs.gov
Appendixes
Information here is presented as PostScript figures only. The files
include sufficient information to permit printing on any standard
PostScript-compatible device; the maps are also compatible with Adobe
Illustrator version 5.0 for the Macintosh.