
UNITED STATES DEPARTMENT OF THE INTERIOR
U.S. GEOLOGICAL SURVEY
Thermodynamic
Data for Modeling Acid Mine Drainage Problems:
Compilation
and Estimation of Data for Selected Soluble Iron-Sulfate Minerals[1]
By
Bruce S.
Hemingway[2],
Robert R. Seal, II[3], and I-Ming
Chou3
Open-File
Report 02-161
Abstract
Enthalpy of formation, Gibbs energy of
formation, and entropy values have been compiled from the literature for the
hydrated ferrous sulfate minerals melanterite, rozenite, and szomolnokite, and
a variety of other hydrated sulfate compounds.
On the basis of this compilation, it appears that there is no evidence
for an excess enthalpy of mixing for sulfate-H2O systems, except for the first H2O molecule of crystallization. The enthalpy and Gibbs energy of formation
of each H2O
molecule of crystallization, except the first, in the iron(II) sulfate - H2O system is ‑295.15 and -238.0
kJ·mol-1, respectively. The
absence of an excess enthalpy of mixing is used as the basis for estimating
thermodynamic values for a variety of ferrous, ferric, and mixed-valence
sulfate salts of relevance to acid-mine drainage systems.
Introduction
Remediation of problems that arise from
release of the components of soluble minerals to ground and surface waters
associated with active and abandoned mines will require geochemical modeling of
the rock-water interactions. Such
models will depend upon an extensive thermodynamic data base. However, thermodynamic data necessary for
such modeling calculations are limited.
This report is a summary of the procedures used to develop a database
for hydrous iron sulfate compounds during the mid-1990s at the U.S. Geological
Survey.
The soluble iron-sulfate minerals are an
important group of phases with regard to acid-mine drainage. These minerals include melanterite FeSO4·7H2O,
rozenite FeSO4·4H2O, szomolnokite FeSO4·H2O,
copiapite Fe2+Fe3+4(SO4)6(OH)2·20H2O,
römerite Fe2+Fe3+2(SO4)4·14H2O,
coquimbite Fe3+2(SO4)3·9H2O,
kornelite Fe3+2(SO4)3·7H2O,
rhomboclase (H3O)Fe3+(SO4)2·3H2O,
voltaite K2Fe2+5Fe3+4(SO4)12·18H2O,
and halotrichite-bilinite Fe2+(Al,Fe3+)2(SO4)4·22H2O,
among others. The compounds have been
shown to play important roles in controlling the extreme mine-drainage
compositions from Iron Mountain mine, California, which reaches pH values less
than zero and total iron concentrations over 100 grams per liter (Nordstrom and
Alpers, 1999). They are also found in
numerous other acid-mine drainage settings (Alpers and others, 1994; Jambor and
others, 2000).
Despite the
importance of the phases, much of the thermodynamic data necessary for such
modeling calculations are not available, or are of questionable quality. For example, considerable uncertainty exists
in the location of the reaction:
FeSO4·4H2O
+ 3H2O(g) = FeSO4·7H2O (1),
(where “g” refers to gas) in terms of
temperature and relative humidity (RH).
Published estimates of the relative humidity associated with this
reaction at 25 °C range from a low of ~15 % (Pribylov, 1969) to a high of ~95 %
(DeKock, 1982) with a distinct clustering around 60 to 80 % (Fig. 1). Results of the ongoing investigations
studying dehydration equilibria between melanterite and rozenite of Chou and
others (1999; 2002) are similar to the results of Malinin and others (1979) and
yield an equilibrium relative humidity of approximately 63 %.
This paper reviews the experimental data for soluble iron- and other sulfate phases and uses these data to construct models for estimating thermodynamic data at 298.15 K and 1 bar for phases for which no experimental data exist. Even though our estimations of thermodynamic properties will greatly enhance the ability to model the geochemistry of acid-mine drainage, they only serve as interim solutions to our inadequate understanding of these systems. A more rigorous treatment must await experimental determination of thermodynamic properties.
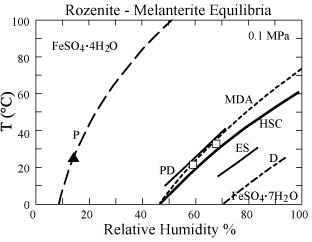
Figure 1. Comparison of estimates for the reaction of
rozenite + H2O to melanterite as a
function of temperature and relative humidity.
Open squares are results of Malinin and others (1979); filled triangle
is result of Pribylov (1969). Sources
of curves are: P, Pribylov (1969); PD, Parkinson and Day (1981); MDA, Malinin
and others (1979); HSC, this study; ES, Ehlers and Stiles (1965); D, DeKock
(1982).
Ferrous
sulfates and their hydrates
Five different ferrous-sulfate hydrate
minerals have been identified and described in the literature. They are, in order of increasing hydration:
szomolnokite FeSO4·H2O, rozenite FeSO4·4H2O,
siderotil FeSO4·5H2O, ferrohexahydrite FeSO4·6H2O,
and melanterite FeSO4·7H2O. In nature, melanterite and rozenite are the most commonly
reported phases, followed by szomolnokite and siderotil (Alpers and others,
1994). Ferrohexahydrite is extremely rare
(Fleischer, 1963). Laboratory studies
of evaporating waters and field observations have defined a general
crystallization sequence of these minerals beginning with melanterite, followed
by rozenite, which in turn is followed by szomolnokite; following szomolnokite
are mixed valence salts followed by ferric salts (Buurman, 1975; Nordstrom and
Alpers, 1999; Jambor and others, 2000).
Notably absent from the crystallization sequence are ferrohexahydrite
and siderotil. Jambor and Traill (1963)
concluded that siderotil was not a stable phase in the binary (FeSO4
- H2O) system, and required at least 5 mole % of copper substituting
for iron to stablize it relative to melanterite and rozenite. In addition, Ehlers and Stiles (1965) were
able to reverse the transition from melanterite to rozenite at near ambient
temperatures and relative humidities between 70 and 80 % without the appearance
of siderotil and ferrohexahydrite.
Therefore, on the basis of these observations, it would appear that
melanterite, rozenite, and szomolnokite are the stable hydrates in the binary
(FeSO4 - H2O) system at ambient conditions.
The thermodynamic data available for
hydrated ferrous sulfates consist of enthalpy of formation values for
melanterite, szomolnokite (Adami and Kelley, 1963), and rozenite (determined
from the data of Larson and others, 1968), and entropy values for melanterite
(Lyon and Giauque, 1949), rozenite (DeKock, 1982), and szomolnokite (Pribylov,
1969). The Gibbs energy of formation
can be calculated for melanterite, rozenite and szomolnokite, where corrections
have been made to insure internal consistency of ancillary thermodynamic data.
Enthalpy
of formation (DfH°)
In the iron(II) system, there appears to
be little or no excess enthalpy of mixing for H2O and ferrous sulfate (except for the
addition of the first H2O
molecule). If the enthalpy of formation
of szomolnokite (FeSO4·H2O) is subtracted from the equivalent
value for melanterite (FeSO4·7H2O) and divided by 6, the
difference in the number of H2O
molecules of hydration, the resulting value of -295.15 kJ·mol-1 for
each added H2O
molecule may be used to estimate the value for the enthalpy of formation of rozenite
(FeSO4·4H2O). The
value of -295.15 kJ·mol-1 compares favorably with the value of ‑294.8
kJ·mol-1 derived from data compiled for numerous hydrated sulfate
salts relative to monohydrates and the value of -304.2 kJ·mol-1 derived from data compiled for
numerous hydrated sulfate salts relative to anhydrous sulfates, as described
below. The thermodynamic data used in
these calculations are from Wagman and others (1982) unless otherwise stated. The value estimated by the summation of the
enthalpy of formation of szomolnokite and 3 times the value derived above for
the contribution of one H2O
molecule is –2,129.1 kJ·mol-1 compared with the measured value for
rozenite of –2,129.2 kJ·mol-1.
Using this model, we estimate the enthalpy of formation of ferrohexahydrite
(FeSO4·6H2O) and siderotil (FeSO4·5H2O)
as -2,719.3 and -2,424.3 kJ·mol-1, respectively.
Hydrated sulfate minerals of other group
VIIIA and adjacent group VIIA, IB and IIB elements of the Periodic Chart show
(1) a similar value to that derived for the iron-sulfate hydrates for the
contribution of H2O
to the enthalpy of formation, and (2) support for the model of no excess
enthalpy of mixing associated with hydration for the metal sulfate- H2O system as limited above (Figs. 2 and
3). For cobalt (7 hydrate and 6
hydrate), nickel (7 hydrate and 4 hydrate), manganese (7 hydrate and 4
hydrate), copper (5 hydrate and 1 hydrate), zinc (7 hydrate and 1 hydrate), and
cadmium (8/3 hydrate and 1 hydrate) hydrated sulfates, the estimated
contributions for one H2O
are -296.33, -290.74, ‑293.73, ‑298.45, -295.54, and -293.91 kJ·mol-1,
respectively. Using these values, we
may estimate the enthalpies of formation of other hydrates of these cations and
compare the estimated and experimental values.
For CoSO4·H2O, CuSO4·3H2O,
FeSO4·4H2O, MnSO4··H2O, MnSO4·5H2O,
NiSO4·6H2O, and ZnSO4·6H2O, the
estimated and measured values for the enthalpy of formation are summarized in
Table 1. The differences between the
estimated and experimental values are all well within experimental error and
thus support this model for the estimation of enthalpy values.
The finding that there is no excess
enthalpy of mixing in the metal sulfate- H2O system is important because many of the
phases found in nature will deviate in H2O content from that given for the
end-member. This model justifies
calculation of the enthalpy of formation of such materials with non-integer
values for the H2O
content. In further support of this
hypothesis, we may calculate the enthalpy contribution of one H2O
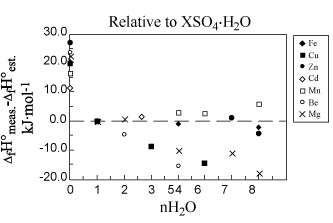
Figure 2. Deviation of estimated enthalpy of formation
values (DfH°est.) from
measured values (DfH°meas.) for
metal-sulfate salts relative to the value for XSO4·H2O. Calculations were made assuming an enthalpy
contribution for each H2O molecule of -294.8
kJ·mol‑1. Note that
data for Be and Mg salts (group IIA) were not used to generate the values for
the enthalpy contribution of additional H2O
molecules because of greater structural differences of these compounds relative
to the groups VIIA, VIIIA, IB, and IIB salts (see text).
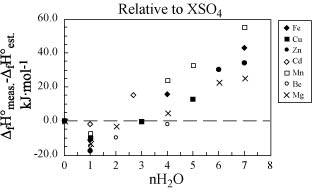
Figure 3. Deviation of
estimated enthalpy of formation values (DfH°est.) from measured values (DfH°meas.) for
metal-sulfate salts relative to the value for XSO4. Calculations
were made assuming an enthalpy contribution for each H2O molecule of ‑304.2 kJ·mol‑1.
The distinct slope in the data is due to the difference between the enthalpy
contribution of the first H2O molecule of
crystallization relative to that of subsequent H2O molecules of crystallization (see text). Note that data for Be and Mg salts (group
IIA) were not used to generate the values for the enthalpy contribution of
additional H2O molecules because of
greater structural differences of these compounds relative to the groups VIIA,
VIIIA, IB, and IIB salts (see text).
Table 1. Comparison of measured and estimated
enthalpy of formation values for selected metal sulfates relative to the
elements and ideal O2, S2, and H2 gases at 298.15 K and 1 bar.
Formula
|
Enthalpy
of formation from elements
|
|
|---|---|---|
|
|
kJ•mol-1
|
|
|
|
Measured*
|
Estimated
|
FeSO4·4H2O
|
-2,129.2
|
-2,129.1
|
CoSO4·H2O
|
-1,199.6
|
-1,201.9
|
NiSO4·6H2O
|
-2,682.8
|
-2,685.6
|
MnSO4·H2O
|
-1,376.5
|
-1,376.9
|
MnSO4·5H2O
|
-2,553.1
|
-2,551.8
|
CuSO4·3H2O
|
-1,684.3
|
-1,682.7
|
ZnSO4·6H2O
|
-2,777.5
|
-2,782.2
|
*All data are from Wagman
and others (1982), except for the datum for CoSO4·H2O,
which is from Goldberg and others (1966), and the datum for FeSO4·4H2O,
which is from Larson and others (1968).
molecule from the difference between the
enthalpies of formation of UO2SO4·3H2O and UO2SO4·1H2O,
which yields -300.95 kJ·mol-1 per H2O.
We use this value to calculate the enthalpies of formation of UO2SO4·3.5H2O
and UO2SO4·2.5H2O and compare them to the
experimental values given for these phases: -2,906.3 (estimated) and -2,900.8
(measured), and -2,603.8 (estimated) and -2,607.1 (measured) kJ·mol-1,
respectively. Again, the differences
between the estimated and experimental values are within the experimental
error.
The enthalpy contribution, except for the
first H2O
molecule, is different for each chemical system, that is, for Fe versus Mn or
Cu. The differences are small and
within the experimental error of such calculations, which suggests that there
will not be large enthalpies of mixing for the H2O component as trace elements substitute
for the end-member cations. The average
value for the contribution of one H2O to the enthalpy of formation of the group VIIA, VIIIA, IB,
and IIB hydrated sulfates is -294.8 ± 2.4 kJ·mol‑1. For the groups IA, IIA, IIIA, IVA and VA
hydrated sulfates (sulfates of Be, Ca, Mg, La, Zr, and mixed K-Mg, K-Zn, K-Cu,
K-Al, Na-Al, and Na-Zn), the value appears to be somewhat larger, about -298 to
-300 kJ·mol-1 probably due to greater structural differences. There are not sufficient data at this time
to determine the value for the group VIA hydrated sulfates, but data from (Cr(H2O)6)2(SO4)3·nH2O
phases suggest a value close to the group VIIIA value.
The enthalpy contribution of the first H2O molecule also varies with the cation,
but again the calculated values are in agreement within experimental
error. Using data from Fe, Mn, Co, Cu,
Cd, and Zn sulfate and sulfate hydrate, the average enthalpy contribution for
the first H2O
molecule is -313.8 ± 5.7 kJ·mol‑1. This value is more negative by 19 kJ·mol-1 than the
average value for additional H2O
molecules of hydration. Thus, when
considered in terms of hydration from an anhydrous salt, the average enthalpy
contribution per H2O
molecule is -304.2 ± 7.9 kJ·mol‑1, which has a larger
uncertainty than that based on hydration of a singly hydrated salt (-294.8 ±
2.4 kJ·mol-1). However, for
cases where an experimentally determined value for a singly hydrated salt is
lacking, this value may be sufficient as a first-order approximation.
Gibbs
energy of formation (DfG°)
The Gibbs energy of formation is the primary thermodynamic quantity needed for modeling rock-water interactions, when solubility data are not available. Few values of the Gibbs energy of formation have been determined for the soluble iron sulfates. The procedure used to estimate the Gibbs energies listed in Table 2 are discussed below.
The Gibbs
energy of formation is related to the enthalpy of formation through the
following thermodynamic equality:
DfG° = DfH° - TDfS° (2),
where DfS° is the entropy of formation at
temperature T in kelvins. It has been
standard practice to add a constant amount for the entropy contribution of each
H2O to the value
of the anhydrous metal sulfate to estimate the entropy of the higher hydrates
for which no experimental data are available (e.g., DeKock, 1982, or Latimer,
1952). This would suggest that there is
little or no excess Gibbs energy of mixing for the metal sulfate-H2O system, except for the first H2O molecule.
Table 2. Thermodynamic properties for selected soluble
iron sulfates relative to the elements and ideal O2, S2,
and H2 gases at 298.15 K and 1
bar.
Phase
|
Formula
|
Formation from
elements
|
References*
|
|||
|---|---|---|---|---|---|---|
|
|
|
Entropy
|
Enthalpy
|
Gibbs Energy
|
S
|
H/G
|
|
|
|
J•mol-1•K-1 |
kJ•mol-1 |
kJ•mol-1 |
|
|
Aqueous species |
|
|
|
|
|
|
|
Fe2+ |
Fe2+ |
101.6 |
-90.0 |
-90.5 |
1 |
1 |
|
Fe3+ |
Fe3+ |
278.4 |
-49.0 |
-16.28 |
1 |
1 |
|
H2O |
H2O |
188.8 |
-241.8 |
-228.6 |
4 |
4 |
Ferrous sulfates |
|
|
|
|
|
|
|
FeSO4 |
FeSO4 |
120.96 |
-932.2 |
-828.3 |
9 |
5 |
|
Szomolnokite |
FeSO4•H2O |
157.7 |
-1,243.69 |
-1,081.2 |
8 |
6 |
|
Rozenite |
FeSO4•4H2O |
282.4 |
-2,129.2 |
-1,795.2 |
10 |
10 |
|
Siderotil |
FeSO4•5H2O |
323.6 |
-2,424.3 |
-2,033.9 |
10 |
10 |
|
Ferrohexahydrite |
FeSO4•6H2O |
368.0 |
-2,719.4 |
-2,271.9 |
10 |
10 |
|
Melanterite |
FeSO4•7H2O |
409.2 |
-3,012.6 |
-2,507.75 |
7 |
1 |
|
Halotrichite |
FeAl2(SO4)4•22H2O |
1,166 |
-11,041 |
-9,306 |
10 |
10 |
Ferric
sulfates
|
|
|
|
|
|
|
|
Fe2(SO4)3 |
Fe2(SO4)3 |
282.8 |
-2,581.9 |
-2,254.4 |
2 |
3 |
|
Kornelite |
Fe2(SO4)3•7H2O |
590.6 |
-4,692.2 |
-3,793.7 |
10 |
10 |
|
Coquimbite |
Fe2(SO4)3•9H2O |
670.1 |
-5,288.2 |
-4,250.6 |
10 |
10 |
|
Ferricopiapite |
Fe5(SO4)6O(OH)·20H2O |
1,396 |
-11,767 |
-9,899 |
10 |
10 |
Mixed ferrous-ferric sulfates |
|
|
|
|
|
|
|
Copiapite |
Fe+2Fe4(SO4)6(OH)2·20H2O |
1,444 |
-11,824 |
-9,971 |
10 |
10 |
|
Römerite |
Fe+2Fe2(SO4)4·14H2O |
943 |
-7,730 |
-6,486 |
10 |
10 |
|
Bilinite |
Fe+2Fe2(SO4)4·22H2O |
1,243 |
-10,121 |
-8,410 |
10 |
10 |
|
Voltaite |
K2Fe+25Fe4(SO4)12·18H2O |
1,959 |
-16,860 |
-14,499 |
10 |
10 |
*References: 1. Parker and Khodakovskii (1995); 2. Barany and Adami
(1965); 3. Pankratz and Weller (1969); 4. Cox and others (1989); 5. Adami and
Kelly (1963); 6. Larson and others (1968); 7. Lyon and Giauque (1949); 8.
DeKock (1982); 9. Pribylov (1969); 10. This study.
The available thermodynamic data for the
ferrous sulfate hydrates are sufficient to develop a model for the contribution
of each H2O
molecule to the Gibbs energy of formation in nearly the same detail used in the
enthalpy of formation calculations.
Four Gibbs energy of formation values are available for use in
estimating the contribution of each H2O molecule. The
difference in the Gibbs energies of formation of the anhydrous sulfate and the
heptahydrate (7) divided by the number of H2O molecules yields a value of -241.3
kJ·mol-1 for the contribution of one H2O molecule. This value must be modified.
It was shown above that the enthalpy contribution of the first H2O molecule is larger than that for
additional H2O
molecules. In the ferrous sulfate
system, the difference was 20.1 kJ·mol-1 in the enthalpy of
formation. This difference divided by 7
is the estimated correction factor required above and yields a value of -238.4
kJ·mol‑1 for the contribution of H2O molecules greater than the first to the
Gibbs energy. The Gibbs energy of
formation for melanterite is based upon the enthalpy of formation reported by
Adami and Kelley (1963) and the entropy reported by Lyon and Giauque (1949). The measurements of the heat capacity of
melanterite between 1 and 300 K (Lyon and Giauque, 1949) fully account for the
magnetic heat capacity expected for Fe2+ and thus for the magnetic
entropy.
We may also calculate the contribution of
one H2O molecule
to the Gibbs energy of formation by dividing the difference in the Gibbs energy
values of rozenite and szomolnokite by 3.
This calculation yields ‑238.0 kJ·mol-1 for the
contribution of one H2O
molecule, in good agreement with the value calculated above. This value was used to calculate the
estimated Gibbs energy values listed in Table 2 beginning with the experimental
Gibbs energy value for melanterite. The
values calculated for the Gibbs energy of formation by this method may be
compared with the experimental values, respectively, for rozenite (‑1,795.2 vs. ‑1,795.9 kJ·mol-1) and
szomolnokite (-1,081.2 vs. -1,081.9 vs. kJ·mol-1).
In support of the value just selected, we
may examine the values for the contribution of additional H2O molecules to the Gibbs energy in other
hydrated metal sulfate systems. Using
the same phases as listed above in the enthalpy calculations unless otherwise
stated, for Co, Ni (7 hydrate and 6 hydrate), Zn, Cu, and Cd sulfate hydrates,
we calculate -238.06, -237.22, -238.45, ‑240.41, and -237.85 kJ·mol-1,
respectively, for the contribution of one additional H2O molecule. The average of these values is -238.4 kJ·mol-1. This value is in obviously good agreement
with the values calculated above for the ferrous sulfate hydrates.
It should be noted that this estimation scheme empirically describes the available data, but in the strictest sense, does not satisfy the thermodynamic properties of ideal mixtures. For ideal mixtures, the Gibbs energy of mixing is described by the relationship:
DG ideal mixing = RT
![]() (3),
(3),
where R = 8.3144 J·mol-1·K-1
and X is the mole fraction in the binary FeSO4·(H2O·6o)
- FeSO4·7H2O. For
rozenite (X = 0.5), the DG ideal mixing should be -1.7
kJ·mol-1 at 298.15 K as opposed to a value of -0.725 kJ·mol-1
based on the data in Table 2 (Fig. 4).
The uncertainty in the measured DfG° value is at least ± 1.5 kJ·mol-1. Therefore, the measured value is within the
limits of its uncertainty with respect to ideal mixing assuming that the values
for the end members are accurate.
Without more data on the Gibbs energy of intermediate hydrates in
various systems, the present study chooses to retain the additive scheme
described above because of its success in empirically describing the available
data. However, the reader is urged to
carefully scrutinize equilibria calculated using these estimates to insure
consistency with natural assemblages and other constraints.
![Figure 4. Variation of the ideal Gibbs energy of mixing with mole fraction of melanterite along the join FeSO4·(H2O·6[hole]) (szomolnokite) - FeSO4·7H2O (melanterite) at 298.15K](./OF02-161_files/figure4.jpg)
Figure 4. Variation of
the ideal Gibbs energy of mixing with mole fraction of melanterite along the
join FeSO4·(H2O·6o)
(szomolnokite) - FeSO4·7H2O (melanterite) at
298.15K. Solid curve represents ideal
mixing calculated on the basis of equation 2.
Solid circles represent experimentally determined values for szomolnokite
(X = 0.0), rozenite (X = 0.5), and melanterite (X = 1.0). Note that the uncertainty on the DfG° value for rozenite is
at least ± 1.5 kJ·mol-1 and is indicated by the vertical bracket.
Entropy
(S°)
The entropy of the ferrous sulfate phases listed in Table 2 may be estimated from the equality given above and the values for the Gibbs energy and enthalpy of formation. It should be noted that these values should be used with great care. Because the entropy derived in this manner is based upon the small difference between two large numbers each with an associated uncertainty, the uncertainty in the entropy is an order of magnitude larger than that for the Gibbs energy or enthalpy of formation. This approach is followed to maintain internal consistency of the thermodynamic data listed in Table 2.
Ferrous
aluminum sulfate hydrate
The enthalpy (-11,008.3 kJ·mol-1)
and Gibbs energy (-9,277.6 kJ·mol-1) of formation of halotrichite
FeAl2(SO4)4·22H2O are estimated
from the summation of the properties of Al2(SO4)3·6H2O,
melanterite, and the values calculated above for the contribution of 9
additional H2O
molecules. The enthalpy of formation of
halotrichite (-11,008.1 kJ·mol-1) was also calculated from the
summation of the enthalpy of Al2(SO4)3·18H2O
(alunogen?) plus that for rozenite. The
two values estimated for the enthalpy of formation of halotrichite are derived
from two different sets of data and the results agree to better than 0.1
percent. However, the correct formula
for alunogen appears to have 17 H2O molecules of crystallization and not 18, and the value
listed by Wagman and others (1982) for alunogen is an estimate taken from
Kelley and others (1946). The
properties of FeAl2O4 were compared with the summation of
the properties of Al2O3 plus fictive FeO (Robie and
Hemingway, 1995). Both the enthalpy and
Gibbs energies of formation estimated for FeAl2O4 by the
summation method were low by about 0.2 percent. The differences between the measured and estimated values for the
enthalpy and Gibbs energy of formation increases as the difference in mineral
structure increases, for example, 1.6 percent for fayalite vs. the oxides FeO
and SiO2. Chen (1975) has
proposed a method for estimating thermodynamic properties based on a knowledge
of these interrelationships (e.g., Hemingway and Sposito, 1996, p. 97). A factor (0.997) was used to adjust the
estimated values for the enthalpy and Gibbs energy of formation because the
reference materials are close in chemistry and structure in terms of the model
used by Chen (1975). The entropy of
halotrichite is calculated from the equality given above and the values
estimated here. The estimated value for
the entropy of halotrichite derived from the adjusted values for the enthalpy
of formation and the Gibbs energy is about 6 percent smaller than the summation
of entropies following the reaction described above. The entropy follows a similar trend to the properties described
in the Chen (1975) model. The value of
6 percent will be used in calculations discussed below to estimate the
correction necessary when using a summation approach to estimate the entropy of
the hydrated sulfates.
Ferric
sulfates and their hydrates
Thermodynamic data for ferric sulfate
phases are very limited. Wagman and
others (1982) gave a value for the enthalpy of formation of anhydrous Fe2(SO4)3,
‑2,581.5 kJ·mol-1, that is based on the calorimetric study by
Barany and Adami (1965). The heat
capacity of anhydrous ferric sulfate between 50 and 300 K was reported by
Pankratz and Weller (1969). Using these
data, DeKock (1982) calculated the Gibbs energy of ferric sulfate. The Gibbs energy calculated by DeKock (1982)
will be incorrect because the entropy is missing 14.9 J·mol-1·K-1
in magnetic entropy per Fe3+ in the compound (see for example,
Gopal, 1966). Cooke and others (1956)
have shown that the magnetic heat capacity anomaly that can be expected for
salts of Fe3+ can occur at very low temperatures. In the case of Fe(NH3CH3)(SO4)2·12H2O,
a good model for the phases examined here, the maximum in the heat capacity
anomaly arising from the magnetic contribution of Stark splitting in Fe3+
was seen at 0.33 K. They also concluded
that the total magnetic heat capacity, and thus the magnetic entropy, could be
accounted for in the measurements they made between about 0.2 and 20 K. The estimated entropy reported by Pankratz
and Weller (1969) for ferric sulfate did not include a contribution for the
magnetic entropy of Fe3+.
The entropy for ferric sulfate is 282.6 J·mol-1·K-1
(Pankratz and Weller, 1969) plus 2 times 14.9 J·mol-1·K-1
or 312.4 J·mol-1·K-1, and the Gibbs energy of formation
is -2,263.0 kJ·mol-1.
The values derived in the preceding
paragraph represent the reference values upon which the estimated values for
the ferric sulfate hydrate phases reported in Table 2 are based. The difference in the enthalpies of
formation of La2(SO4)3 and La2(SO4)3·9H2O
is 2,706.7 kJ·mol-1 or about 300.7 kJ·mol-1 per H2O molecule. This value is used to estimate the enthalpy of formation of
coquimbite (Fe2(SO4)3·9H2O). Assuming that the first H2O molecule contributes more than
subsequent H2O
molecules and following the model discussed above, we estimate the contribution
of the additional H2O
molecules to be -298 kJ·mol-1 of H2O and use this value to calculate the
enthalpy of formation of kornelite (7 hydrate) from the value estimated for
coquimbite. The entropies of coquimbite
and kornelite are estimated following the procedure described by DeKock (1982)
in which the contribution of each H2O molecule is taken as 39.75 J·mol-1·K-1. The Gibbs energies of the two phases are
calculated from these data and the equality given above.
The enthalpy of formation and entropy for
ferricopiapite were estimated from the summation of these properties for 2
coquimbite, goethite, and 2 H2O
(H2O value as derived
above). The entropy of goethite (60.38
J·mol-1·K-1) was taken from Khodakovsky and others
(1991). These values were adjusted in
accordance with the differences noted above for summation data.
Mixed
ferrous and ferric sulfates
There are few thermodynamic data for
mixed ferrous and ferric compounds so the model used to estimate the
thermodynamic data for this type of phase is similar to that used for
halotrichite. The mixed iron sulfate
hydrate phases are assumed to have slightly larger (about 0.3 percent)
enthalpies of formation than the summation of values for the ferrous and ferric
sulfate hydrates. The enthalpy of
formation for bilinite and römerite are calculated from the sum of the
properties of kornelite and melanterite (and plus 8 H2O molecules for bilinite). The entropies are calculated from the same
summation procedure and corrected by 6 percent as noted above. The Gibbs energies are calculated from these
values using the equality given above.
The values for copiapite are estimated using the same model as for the
other mixed valence iron sulfates, but a correction is made for the
substitution of (OH)2 for SO4. The correction is derived from the difference in the properties
of FeSO4 and Fe(OH)2 (359.4 kJ·mol-1 and 19.5
J·mol-1·K-1, respectively, for the enthalpy and
entropy). The values for voltaite are
derived from the summations and suggested adjustments of the enthalpy of
formation and entropy for 5 szomolnokite, 2 kornelite, K2SO4,
and -1 H2O.
Estimated
uncertainties
The uncertainties estimated for the enthalpy and Gibbs energy of formation values, relative to the elements, derived from the model calculations used in this study are between 0.3 and 0.7 percent, with the values for the ferrous and ferric phases at the low end and those of the mixed phases at the high end.
References
Adami, L.H. and Kelley, K.K., 1963, Heats
of formation of two crystalline hydrates of ferrous sulfate: U.S. Bur. Mines
Rept. Inv. 6260, 7 p.
Alpers, C.N., Blowes, D.W., Nordstrom,
D.K., and Jambor, J.L., 1994, Secondary minerals and acid mine-water chemistry,
in The Environmental Geochemistry of
Sulfide Mine-wastes, Blowes, D.W. and Jambor, J.L., eds., Mineral. Assoc.
Canada Short Course Notes, v. 22, p. 249-269.
Barany, R., and Adami, L.H., 1965, Heats
of formation of anhydrous ferric sulfate and indium sulfate: U.S. Bur. Mines
Rept. Invest. 6687, 8 p.
Buurman, P., 1975, In vitro weathering products of pyrite: Geologie en Mijnvouw, v.
54, p. 101-105.
Chen, C.H., 1975, A method of estimation
of standard free energies of formation of silicate minerals: Am. J. Sci., v.
275, p. 801-817.
Chou, I-M., Seal, R.R., II, and
Hemingway, B.S., 1999, Determination of melanterite-rozenite and
chalcanthite-bonattite equilibria by the humidity buffer technique: In Ninth Annual V.M. Goldschmidt Conference,
LPI Contribution No. 971, Lunar and Planetary Science Institute, Houston, p.
54-55.
Chou, I-M., Seal, R.R., II, and
Hemingway, B.S., 2002, Determination of melanterite-rozenite and
chalcanthite-bonattite equilibria by the humidity measurements at 0.1 MPa:
American Mineralogist, v. 87 , p. 108-114.
Cooke, A.H., Meyer, H., and Wolf, W.P.,
1956, Thermal and magnetic properties of ferric methylammonium sulphate: Proc.
Roy. Soc. London, v. A237, p. 404-412.
Cox, J.D., Wagman, D.D., and Medvedev,
V.A., 1989, CODATA key values for thermodynamics: Hemisphere, New York, 271 p.
DeKock, C.W., 1982, Thermodynamic
properties of selected transition metal sulfates and their hydrates: U.S. Bur.
Mines Info. Circ. 8910, 45 p.
Ehlers, E.G., and Stiles, D.V., 1965,
Melanterite-rozenite equilibrium: Amer. Mineral., v. 50, p. 1457-1461.
Fleischer, M., 1963, New mineral names:
Amer. Mineral., v. 48, p. 433.
Goldberg, R.N., Riddell, R.G., Wingard,
M.R., Hopkins, H.P., Wulff, C.A., and Hepler, L.G., 1966, Thermochemistry of
cobalt sulfate and hydrates of cobalt and nickel sulfates: J. Phys. Chem., v.
70, p. 706-710.
Gopal, E.S.R., 1966, Specific heats at
low temperatures: Plenum Press, New York, 240 p.
Hemingway, B.S. and Sposito, G., 1996,
Chapter 3. Inorganic aluminum-bearing solid phases: in The Environmental
Chemistry of Aluminum, G. Sposito, ed., CRC Press, Boca Raton, FL, p. 81-116.
Jambor, J.L., and Traill, R.J., 1963, On
rozenite and siderotil: Can. Mineral., v. 7, p. 751-763.
Jambor,
J.L., Nordstrom, D.K., and Alpers, C.N., 2000, Metal-sulfate salts from sulfide
mineral oxidation: In: Alpers, C.N., Jambor, J.L., and Nordstrom, D.K., eds.,
Sulfate minerals: crystallography, geochemistry, and environmental
significance, Reviews in Mineralogy and Geochemistry, v. 40, p. 305-350.
Kelley, K.K., Shomate, C.H., Young, F.E.,
Naylor, B.F., Salo, A.E., and Huffman, E.H., 1946, Thermodynamic properties of
ammonium and potassium alums and related substances, with reference to
extraction of alumina from clay and alunite: U.S Bureau of Mines Technical
Paper 688, 104 p.
Khodakovsky, I.L., Westrum, E.F., Jr.,
and Hemingway, B.S., eds., 1991, International Geothermodynamic Tables.
Guidelines and Set of Prototype Tables: CODATA Task Group on Geothermodynamic
Data, BETA-Review copy available from editors, 256 p.
Larson, J.W., Cerutti, P., Garber, H.K.,
and Hepler, L.G., 1968, Electrode potentials and thermodynamic data for aqueous
ions. Copper, zinc, cadmium, iron, cobalt, and nickel: J. Phys. Chem., v. 72,
p. 2902-2907.
Latimer, W.M., 1952, The oxidation states
of the elements and their potentials in aqueous solutions, Prentice Hall, New
York, 392 p.
Lyon, D.N. and Giauque, W.F., 1949, Magnetism and the third law of
thermodynamics. Magnetic properties of ferrous sulfate heptahydrate from 1 to
20° K. Heat capacity from 1 to 300° K: J. Amer. Chem. Soc., v. 71, p.
1647-1657.
Malinin, A.A., Drakin, S.I., and
Ankudimov, A.G., 1979, Equilibrium dehydration pressures of salt crystal
hydrates: Russian Journal of Physical Chemistry, v. 53, p. 1332-1333.
Nordstrom, D.K. and Alpers, C.N., 1999,
Negative pH, efflorescent mineralogy, and consequences for environmental
restoration at the Iron Mountain Superfund site, California: Proc. Natl. Acad.
Sci. USA, v. 96, p. 3455-3462.
Pankratz, L.B. and Weller, W.W., 1969,
Thermodynamic data for ferric sulfate and indium sulfate: U.S. Bur. Mines Rept.
Invest. 7280, 9 p.
Parker, V.B., and Khodakovskii, I.L.,
1995, Thermodynamic properties of the aqueous ions (2+
and 3+)
of iron and the key compounds of iron: J. Phys. Chem. Ref. Data, v. 24, no. 5,
p. 1699-1745.
Parkinson,
K.J., and Day, W., 1981, Water vapor calibration using salt hydrate
transitions. Journal of Experimental Botany, v. 32, p. 411-418.
Pribylov, K.B., 1969, Determination of the
thermal effects of the dehydration of FeSO4·7H2O: Russ.
J. Inorg. Chem., v. 14, p. 168-169.
Robie, R.A. and Hemingway, B.S., 1995,
Thermodynamic properties of minerals and related substances at 298.15 K and 1
bar (105 Pascals) pressure and at higher temperatures: U.S. Geol.
Surv. Bull. 2131, 461 p.
Wagman, D.D., Evans, W.H., Parker, V.B.,
Schumm, R.H., Halow, I., Bailey, S.M., Churney, K.L., and Nuttall, R.L., 1982,
The NBS tables of chemical thermodynamic properties. Selected values for inorganic and C1 and C2
organic substances in SI units: Jour. Phys. Chem. Ref Data, 11, Sup. 2.