The fine grained (minus-100 mesh, or minus-150-micron) fraction of the stream sediments was analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) utilizing both a total digestion procedure and a partial-digestion leach procedure (2M HCl-1%H2O2) on separate splits of each sample. The total decomposition procedure utilizes a combination of hydrochloric, nitric, perchloric and hydrofluoric acids, and is effective in dissolving most minerals, including silicates, oxides, and sulfides. Some resistant or refractory minerals such as zircon, chromite and selected tin oxide minerals are only partially attacked with this procedure (Crock and others, 1983 Briggs, 1996). Previous investigations using a variety of materials support the completeness of this digestion (Church, 1981 Church and others, 1987 Wilson and others, 1994). The partial-digestion leach procedure used involves an extraction performed on the samples using a solution of warm 2M HCl-1%H2O2 (appendix III of Church and others, 1993). This leaching solution dissolves hydrous amorphous iron- and manganese-oxide minerals, as well as some crystalline iron- and manganese-oxides. The procedure we used differed slightly from that described. We used a 2-g sample in 15 mL of reagent. The samples were placed in 90 mL Teflon FEP jars, sealed, and placed in the waterbath at 50o C for three hours to ensure complete removal of the iron- and manganese-oxide coatings from the sediment grains.
Selenium was determined using hydride generation-atomic absorbtion spectrophotometry (Hg-AAS), as was arsenic for those samples whose ICP-AES values were less than 20 ppm. Samples were digested using a combination of nitric, hydrochloric, perchloric, sulfuric and hydrofluoric acids in open teflon vessels (Hageman and Welsch 1996), resulting in total dissolution. The arsenic and selenium in the solutions were reduced to the +3 and +4 states respectively, using a potassium iodide-ascorbic acid solution, after which the respective hydrides were generated by the addition of and mixing with sodium borohydride. The gaseous hydrides were introduced into a heated quartz tube in the optical path of an atomic absorbtion spectrophotometer and analyzed.
Mercury was determined using a cold vapor-atomic absorbtion spectrophotometric method (CV-AAS). Samples were digested using concentrated nitric acid, with the addition of sodium dichromate (O'Leary and others, 1996). The solutions were then introduced into a continuous flow manifold and mixed with air and a sodium chloride-hydroxylamine hydrochloride-sulfuric acid solution. The mercury +2 was then reduced to elemental mercury using stannous chloride, and the vapor introduced into the optical path of an atomic absorbtion spectrophotometer and analyzed.
Samples were randomized and submitted to the laboratory as blind samples. Analytical precision and accuracy of the methods (quality control) were assessed by analyzing blind standard reference materials (SRMs) and a replicate sample (a split of a sample, submitted as two separate samples) with each analytical set. These data are given in table A-II for the SRM samples in the data tables in appendices V and VI for replicate analyses of blind duplicate samples. The SRMs were NIST-2704, NIST-2709, NIST-2710, and NIST-2711, available from the National Institute of Standards and Technology (NIST, 1993a, 1993b, 1993c and 1993d). The results for the total-digestion ICP-AES, and for arsenic, selenium and mercury compare favorably with previous work (Church and others, 1993, 1994, and 1995), and with the published NIST values.
The lead-isotopic data on the sediment samples were measured using the 2M HCl-1%H2O2 digestion solution following the procedure described in appendix IV of Church and others (1993). The lead was separated from solution in the HBr medium and the lead-isotopic ratios determined on a 68o sector, 12-inch radius, solid-source, thermal ionization NBS mass spectrometer. Analytical precision, based upon replicate analyses of NIST SRM materials is better than 0.1% per mass unit for the reported lead-isotopic ratios. The analytical results are reported in appendix V.
Legally and in many standard methods, a dissolved-metal concentration is defined as the concentration that passes through a 0.45-micrometers (mm) filter (Horowitz and others, 1996a). Hydrous iron oxides can be on the order of 0.001 mm when they initially form in stream water. However, such small particles rapidly aggregate, forming a continuous size range from 0.001 mm to greater than 1 mm (Stumm and Morgan, 1996). Thus, for iron-rich streams affected by mine drainage, 0.45 mm is neither an effective nor a natural break for the distinction of dissolved and particulate concentrations. These artifacts confound the sampling procedure and skew the data introducing sampling errors that make it difficult to make a meaningful geochemical interpretation (Shiller and Taylor, 1996 Horowitz and others, 1996b). Many standard methods of water sampling protocol rely on filtration to obtain "dissolved" concentrations. The accepted standard protocol for many regulatory statutes relies on filtration of water samples at an arbitrary breaking point of 0.45 mm. Horowitz and others (1996a) described numerous problems that result from filtration with standard 0.45-mm filters. A recent study in the upper Arkansas River basin demonstrated that iron colloids can be separated from river water and analyzed for their metal content using ultrafiltration (Kimball and others, 1995). The methods developed in that study have been followed here with only a slight change in the approach to separate and analyze the colloidal metal concentrations. In this study tangential-flow ultrafiltration through a membrane with an effective pore size less than 0.001 mm was used to differentiate between dissolved and particulate metal fractions (Hernandez and Stallard, 1988 Moran and Moore, 1989 Kimball and others, 1995).
The procedure to distinguish concentrations of metals in water from concentrations in colloidal particles is summarized in figure A-II. The integrated sample was screened to remove sand, gravel, and debris. However, there was very little material greater than 62 mm. Almost the entire load of the Animas River was dissolved and colloidal. A split of the screened sample allowed the determination of a "total recoverable metal concentration." The rest of the sample was used for ultrafiltration to determine two metal concentrations, the "dissolved" concentration in the filtrate less than 0.001 mm, and the "colloidal" concentration from the concentrated colloids in water. Tangential-flow filtration keeps solid material in suspension rather than forcing it against a filter membrane. This allows water to be removed by osmotic pressure across a filter membrane without "packing" the suspended solids onto a filter membrane, which would change the membrane pore size.
Indirect measurement of metal concentrations in colloids was made by calculating the difference between the metal concentrations in an unfiltered, acidified sample (a total recoverable concentration) and the ultrafiltrate sample. The indirect measurement was reliable for iron, manganese, and zinc at each of the sites, but the direct measurement was necessary for cadmium, copper, and lead because of their lower concentrations.
Dissolved and total recoverable metal concentrations were determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES Briggs, 1996). Anions were analyzed in the 0.45-mm filtered, unacidified samples by ion chromatography. Alkalinity was measured by Gran titration of an unfiltered 0.45-mm filtered sample.
The low levels encountered for cadmium, copper, nickel, and lead indicate that either graphite furnace atomic adsorption or ICP Mass Spectrometry should be used for better quantification. The ICP-AES did not allow detection of lead at low levels because the lead concentrations were below the limit of detection.
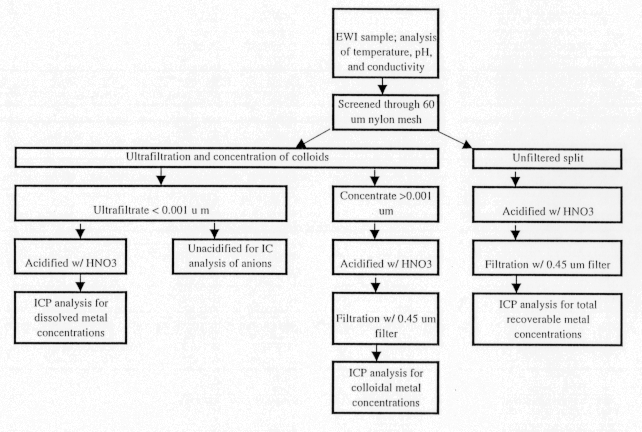 Figure A-II. Diagram showing processing procedures for water and colloid samples.
Figure A-II. Diagram showing processing procedures for water and colloid samples.