
Scientific Investigations Report 2004–5121
Degradation pathways of plant tissue components
Interactions of degradation products
Figure 1. Lignin monomeric units.
Figure 2. Lignin degradation pathways.
Figure 3. Cutin-forming acids.
Figure 4. Model cutin monomeric unit according to Kolattukudy (2001).
Figure 5. Hydrolysable tannin units.
Figure 6. Ellagitannin formation.
Figure 7. Nonhydrolysable tannins.
Figure 9. Formation of anhydro sugars.
Figure 11. Formation of benzotropolone rings according to Haslam (1998).
Figure 12. Auto-oxidation of pyrogallol (Haslam, 2003).
Figure 13. Possible oxidation reactions of thearubigins according to Haslam (2003).
Figure 14. Thearubigin units according to Davies and others (1999).
Figure 15. Formation of anthocyanin pigments (Salas and others, 2003).
Figure 16. Proanthocyanidin oligomers.
Figure 17. Bicyclic and xythylium salt forms of flavene dimers.
Figure 18. Effect of pH on the structure anthocyanins.
Figure 19. First steps in browning reactions.
Natural organic matter (NOM) has been studied for more than 200 years because of its importance in enhancing soil fertility, soil structure, and water-holding capacity and as a carbon sink in the global carbon cycle. Two different types of models have been proposed for NOM: (1) the humic polymer models and (2) the molecular aggregate models. In the humic polymer models, NOM molecules are depicted as large (humic) polymers that have unique chemical structures that are different from those of the precursor plant degradation products. In the molecular aggregate models, NOM is depicted as being composed of molecular aggregates (supramolecular aggregates) of plant degradation products held together by non-covalent bonds. The preponderance of evidence favors the supramolecular aggregate models. These models were developed by studying the properties of NOM extracted from soils and natural waters, and as such, they provide only a very generalized picture of the structure of NOM aggregates in soils and natural waters prior to extraction. A compartmental model, in which the structure of the NOM in each of the compartments is treated separately, should provide a more accurate representation of NOM in soil and sediment systems. The proposed NOM compartments are: (1) partially degraded plant tissue, (2) biomass from microorganisms, (3) organic coatings on mineral grains, (4) pyrolytic carbon, (5) organic precipitates, and (6) dissolved organic matter (DOM) in interstitial water. Within each of these compartments there are NOM supramolecular aggregates that will be dissolved by the solvent systems that are used by researchers for extraction of NOM from soils and sediments. In natural water systems DOM may be considered as existing in two subcompartments: (1) truly dissolved DOM and (2) colloidal DOM.
Soils and sediments are composed of complex mixtures of inorganic and organic components. The organic components of soils (soil organic matter) constitute the largest pool of carbon on the surface of the Earth (González-Pérez and others, 2004). This carbon pool is particularly important because soil properties such as buffering capacity, metal-binding capacity, stability of aggregates of soil particles, water-holding capacity, and sorption of hydrophobic organic compounds are dependent, to a large extent, on the amount of organic matter in a soil. All of these properties, with the exception of the last one, are important in controlling soil fertility.
The maintenance of soil fertility is of paramount importance for the survival of human life on our planet. In the rich, industrialized countries of the world soil fertility is maintained by application of chemical fertilizers that are produced using large amounts of fossil fuels. The costs of chemicals fertilizers are expected to increase because of increases in costs of fossil fuels. The ready availability of the chemical fertilizers has encouraged farmers to employ intensive agricultural practices, such as growing a single crop year after year (monoculture), irrigation, and heavy use of pesticides. These practices oftentimes result in severe erosion and deterioration of soil texture and aggregation (Pimentel and others, 1995; Tilman, 1999).
Mäder and others (2002) showed that organic farming systems are much more sustainable than conventional intensive agriculture. In addition, they found that organic farming reduced energy imput by 34 to 53 percent and pesticide imput by 97 percent. In organic farming practice, nutrients are supplied by the application of manure, green manure, and compost, and by growing legumes. Organic farming results in a marked increase in concentration of organic matter in an agricultural soil. The development of a more refined conceptual model of this organic matter will be the topic of this report. Such a model is necessary in order to be able to optimize sustainable agricultural practices and to better understand the chemical and biochemical reactions that take place in natural water systems.
The pool of NOM in soils is not only an important factor in soil fertility, it also serves as a carbon sink. Increasing this pool not only improves soil fertility, it also reduces the amount of anthropogenic carbon dioxide that is added to the atmosphere. Thus, the active sequestration of carbon in soil can be an important tool in reducing climate change brought by the burning of fossil fuels and plant biomass.
Naturally occurring organic compounds in soils have been studied for more than 200 years because agricultural scientists early recognized the importance of natural organics in enhancing soil fertility, soil structure, and water-holding capacity (Kononova, 1966). The most general term for the natural organic compounds in soils, sediments, and natural waters is natural organic matter (NOM); however, a number of other terms have been applied to different NOM fractions. The most commonly used of these are: humus, humic substances, humic acid, fulvic acid, and humin. The dissolved fraction of the NOM in natural waters is commonly called dissolved organic matter (DOM) or dissolved organic carbon (DOC). Historically, the term humus has been applied to the dark-colored, "rotted" organic matter in soils, and the terms humic acid, fulvic acid, and humin have been used to designate different fractions of humus (Kononova, 1966).
Early workers (see Kononova, 1966, p. 13-45) understood that soil humus arises mainly from the degradation of the dead plant tissue with a lesser contribution from decaying animal remains. Many of these early workers assumed that humus was composed of the end products of synthetic reactions that alter the structures of plant degradation products. Other workers, however, maintained that humus is a complex mixture of plant degradation products. This controversy has persisted. Stevenson (1994) has provided a detailed exposition of the synthetic reaction postulate. He defined soil humic substances as "a series of relatively high-molecular weight, yellow to black colored substances formed by secondary synthesis reactions," and humus as the "total of the organic compounds in soil exclusive of undecayed plant and animal tissues, their 'partial decomposition' products, and the soil biomass." In contrast, a number of workers recently have proposed that humic substances consist mainly of the partial degradation products of plant polymers (Baldock and others, 1992; Hatcher and Spiker, 1988; Piccolo, 2001; Wershaw, 1986; Wershaw, 1994). These workers generally do not explicitly state, however, as Stevenson (1994) does, what NOM compounds are excluded from the category of humic substances; therefore, their definition of humic substances is necessarily incomplete. Most workers would agree that soil NOM consists of humified components (humus or humic substances) and the nonhumified components (unaltered plant components). Those workers who do not accept Stevenson's definitions must, therefore, distinguish between those NOM components that have been altered enough to be called humic substances and those that have not. It is probably useful to exclude completely undecomposed plant and animal tissue and living microorganisms from the definition of humus; however, there is no practical way to exclude "partial decomposition" products from humus isolates, and indeed, it is likely that soil humus consists mainly of such products (Wershaw, 1994; Knicker and others, 1997).
This controversy has led to the development of two different types of models for NOM: (1) the humic polymer model and (2) the molecular aggregate model. The belief that the component molecules of NOM are produced by secondary synthesis reactions from degradation products has led to models in which the NOM molecules are depicted as polymeric species that have distinctive chemical structures that are different from those of starting materials. These models, for want of a better designation, will be called humic polymer models in the following discussion. The proposal that NOM is composed of the partial degradation products of plant polymers has resulted in the development of models in which NOM is composed of molecular aggregates of the degradation products held together by non-covalent bonds (Wershaw, 1994; Piccolo, 2001). Following the usage of Piccolo (2001), these models are designated as supramolecular aggregate models.
In order to resolve this controversy Wershaw (2000) has proposed a paradigm shift in the study of NOM. He stated "* * *that a more fruitful approach to the study of NOM in soils and natural waters than that based on the humic substance paradigm is to study the chemical reactions that the chemical components of plant tissue undergo during and after senescence. That is to say, to concern oneself with the humification process rather than with ill-defined intermediates in the continuum from well-characterized plant components to carbon dioxide." Humification may be considered as a three-step process of (1) degradation of components of plant tissue followed by (2) reassembly of the degradation products into NOM, and (3) the degradation of the NOM formed in step 2. The development, testing, and refinement of conceptual models are essential elements of scientific inquiry.
A good model leads scientists to ask the right questions, that is to say, to design experiments that provide increased insight into the question at hand. At the present time, as pointed out above, there are two very different models for natural organic matter (NOM): (1) the humic polymer model and (2) the molecular aggregate model. Acceptance of one or the other of these models, either explicitly or implicitly, underlies the experimental design of most research in the field.
In this report the likely degradation pathways of the components of plant tissue will be discussed along with the likely mechanisms of interactions of the resulting degradation products in soils and natural waters. An analysis of these mechanisms will be used to develop a compartmental model of NOM. This model is a refinement of a previously proposed aggregate model for NOM (Wershaw, 1994). Techniques for the separation of the NOM compartments will also be described.
The components of plant tissue can undergo three possible types of degradation reactions in natural systems: (1) biotic (enzymatically catalyzed) reactions, (2) pyrolytic reactions, and (3) abiotic reactions exclusive of pyrolytic reactions. In most natural systems, it appears that the degradation of plant tissue is mainly the result of enzymatically catalyzed reactions or sporadic fires that burn plant material. Purely abiotic reactions appear to be less common; although plant components exposed to sunlight are subjected to abiotic photolytic degradation, this degradation often takes place in concert with biodegradation. Zepp and others (1995) pointed out that biodegradation may be accelerated or retarded by increases in solar ultraviolet radiation depending on the properties of each particular environmental system. Abiotic oxidative coupling reactions may also be important in some environments; however, these reactions are often enzymatically catalyzed (Dec and others, 2003). Enzymatic reactions will be considered first followed by pyrolytic reactions and abiotic reactions.
Enzymatic reactions are very substrate specific, and therefore, each chemical structural group of plant components must be dealt with separately. In our discussion of enzymatic degradation, NOM precursors will be divided in two categories: primary metabolites and secondary metabolites. According to Haslam (1995), primary metabolites are those which have "* * *a distinctive role in the life of all organisms* * *. * * *The pathways by which they are synthesised are similar if not identical in all organisms* * *." Secondary metabolites on the other hand, "* * *occur sporadically throughout nature and the question of the role which they, or the processes by which they arise, may play in the economy and welfare of the producing organism is one which excites continued speculation." Haslam (1998) has pointed out that secondary metabolites have three characteristics: (1) structural diversity of the metabolites, (2) accumulation of substantial quantities of the metabolites in plant tissue, and (3) limited taxonomic distribution. Thus the degradation products of primary metabolites will be found in practically all NOM mixtures, whereas those of secondary metabolites will be dependent on the source organisms for each particular NOM.
Biodegradation of primary metabolites is carried out both by the organisms that originally produced the metabolites and by exogenous organisms. For example, carbohydrates and proteins that are produced in plant leaves are degraded during senescence by endogenous enzyme systems to monomeric species that can be stored for use in the following growing season. In addition, heterotrophic, exogenous microorganisms are dependent on primary metabolites produced by plants in their metabolic processes. The enzyme systems that catalyze these endogenous and exogenous reactions are often very similar. The biodegradation of primary metabolite polymers generally involves hydrolytic depolymerization reactions that ultimately produce monomeric species. As these reactions proceed, the average size of the molecules continually decreases until only monomers are left. Primary metabolites also undergo oxidative degradation during cell respiration to produce energy (catabolism). Organic acids produced during catabolism ultimately are utilized in one of the cell respiration cycles such as the citric acid cycle. Size reduction also occurs during oxidative degradation.
Secondary metabolites are more specialized in function than primary metabolites and generally do not possess the structural regularity of primary metabolites. The lack of structural regularity of secondary metabolites causes their biodegradation pathways to be more complex than those of primary metabolites. The degradation of a secondary metabolite does not result in the release of a suite of well-defined monomeric units; in fact, it can lead to the formation of more complex structures. Of particular interest are the plant polyphenols (hydrolysable and nonhydrolysable tannins) and their degradation products that can form non-covalent bonds with proteins and carbohydrates. These are the types of compounds that promote the aggregation of the molecular fragments that constitute NOM.
In addition to NOM components derived from plant biodegradation processes, black carbon (BC), which arises from the burning of plants, should be included as an important constituent of NOM (D.W. Rutherford, U.S. Geological Survey, written commun., 2004). BC provides ion-exchange sites for the binding of plant nutrients thereby improving soil fertility (Glaser and others, 2001; Lehmann and others, 2003). In addition to binding inorganic constituents, BC strongly binds hydrophobic organic compounds; BC has a much higher sorption capacity for these compounds than other NOM components (Karapanagiotti and others, 2000; Yang and Sheng, 2003).
Consideration of the formation processes of NOM in soil systems leads to a model in which NOM is represented as existing in separate interacting compartments. A tentative list of these compartments includes partially degraded plant tissue, biomass from microorganisms, organic coatings on mineral grains, pyrolytic carbon (charcoal), organic precipitates, and DOM in interstitial soil water. Within each of these compartments it is likely that there are degradation products that will form supramolecular aggregates in solution. These degradation products will be dissolved by the solvent systems that are used for extraction of NOM from soils and sediments. At the present time, methods do not exist for separating all of these compartments, but a start has been made. By studying each of these compartments separately it should be possible to develop a much more complete understanding of the properties of NOM in soils.
The discussion in this section illustrates the degradation pathways and the processes that lead to the formation of NOM from degradation products of the organic components of plant tissue; it is not intended to be an exhaustive compendium of all possible groups of plant components. Only the most abundant or reactive groups of compounds will be discussed. It would be especially difficult to provide a comprehensive compendium of the reactions of all of the various types of secondary metabolites. This section is divided into subsections on enzymatic, pyrolytic, and abiotic reactions.
Reactions in biological systems are catalyzed by highly specialized proteins called enzymes. Enzymes are generally substrate-specific, and each enzyme normally catalyzes only a single type of reaction, such as hydrolysis.
Carbohydrates are the most abundant components of plants. They act as both structural and energy-storage components of plants. Cellulose is the main structural component of vascular plants. It generally exists in the plants as crystalline or partially crystalline microfibrils of β-1-4-glucopyranose chains that are relatively resistant to biological degradation. In the native state the microfibrils are present in fiber-like aggregates (Atalla, 1999; Larsson and others, 1999). The microfibrils in these aggregates are bound together by hydrogen bonds between hydroxyl groups and van der Waals interactions between the hydrophobic surfaces of the pyranose rings.
Crystalline native cellulose (cellulose I) is a composite of two different crystalline allomorphs (I∝ and Iβ) that have distinct 13C nuclear magnetic resonance (NMR) spectra (Kono and others, 2002). The preponderance of evidence indicates that the unit cell of the I∝ allomorph consists of a single chain of anhydrocellobiose units, whereas the unit cell of th Iβ allomorph consists of two chains of anhydrocellobiose units (Atalla and VanderHart, 1999; Atalla, 1999). Typically, cellulose aggregates are composed of domains of the two allomorphs and noncrystalline domains. In lower organisms native cellulose fibers occur in a relatively pure state; however, in higher plants the cellulose fibers are generally found in intimate contact with the hemicellulose components of cell walls.
The hemicellulose components are mainly composed of β-1-4-linked pentoses and hexoses; however, some β-1-3-gycosidic bonds also may be present. Some branching of the chains is present in most of the hemicellulose components. The monomeric units of hemicelluloses are: D-xylose, D-mannose, D-galactose, D-glucose, L-arabinose, 4-O-methylglucuronic acid, D-galacturonic acid, and D-glucuronic acid (Pérez and others, 2002). The relative concentrations of these monomers vary in different hemicelluloses. For example, hardwood hemicelluloses are composed mainly of glucuronic acid and xylose units, whereas softwood hemicelluloses are composed mainly of glucose and mannose units. In contrast to cellulose, hemicelluloses are soluble in dilute alkaline solutions. They may be isolated from plant tissue with dilute alkali after the plant pectins have been extracted with aqueous solvents and calcium chelating agents (Gregory and Bolwell, 1999).
Pectins are polysaccharides that contain D-galacturonic acid residues in addition to other glycosyl residues (see Mohnen, 1999). Three groups of pectic polysaccharides have been extensively studied: rhamnogalacturonan I, rhamnogalacturonan II, and homogalacturonan (see Albersheim and others, 1996). They are abundant in the primary walls of plant cells; much lower concentrations of pectins are found in the secondary walls. Albersheim and others (1996) have pointed out that the pectic polysaccharides form networks in the primary cells of plants that are intimately associated with the cellulose and hemicellulose networks in the cells. Pectins are soluble in aqueous solutions; if the pectins are present as calcium salts then chelation of the calcium ions may be necessary. Water-soluble pectin-like gums also are present in the seeds, roots, and bark of some plants.
Pérez and others (2002) have reviewed the biodegradation of cellulose. Cellulose is degraded aerobically by eubacteria and fungi to carbon dioxide and water, and is degraded anaerobically by protozoa and slime molds to methane and water. These organisms secrete a variety of enzymes that attack cellulose in different ways. Cellulases hydrolyze β-1-4-gycosidic bonds; two different types of cellulases have been identified: endoglucanases and cellobiohydrolases (exoglucanases). The endoglucanases hydrolyze internal cellulose bonds; the most susceptible bonds are those in the noncrystalline regions of the cellulose fibers. The cellobiohydrolases attack the ends of the cellulose chains. Both types of enzymes ultimately reduce the cellulose chains to the disaccharide cellobiose. In order for the microorganisms to be able to use the hydrolysis products as an energy source, the cellobiose must be cleaved into two glucose molecules by β-glucosidases. Therefore, in order for microorganisms to effectively use cellulose as an energy source, they must secrete a mixture of endoglucanases, cellobiohydrolases, and β-glucosidases that are stable in the local environment.
Anaerobic degradation has been studied less well than aerobic degradation. Pérez and others (2002) pointed out that cellulose degradation by the strict-anaerobic sporulated bacterium Clostridium thermocellum has been better studied than that caused by any other organism. In the Clostridium thermocellum system the enzymes are packaged in cellulosomes that are attached to the cell surfaces of the organisms. The cellulase enzymes are associated with the protein scafoldin that localizes the various enzymes so that they can more effectively act in concert (Atalla, 1999). The products of cellulolysis pass to the inside of the organisms through fibers that extend from the cellulose to the bacterial cells. Clostridium thermocellum is a thermophilic bacterium; mesophilic cellulolytic anaerobes have been isolated from soils, sediments, composts, and other anaerobic systems. Bacteria, fungi, and protozoa also anaerobically degrade cellulose in the rumina of ruminants.
The hydrolytic degradation of hemicelluloses by hemicellulases produces monomeric saccharides and acetic acid (Pérez and others, 2002). Enzymes specific for the different saccharides and bonds in a given type of hemicellulose are required for the degradation of the particular hemicellulose. For example, if a hemicellulose is made up of chains of β-1-4-linked xylose units, then endo-1-4-β-xylanase is needed to cleave the chains into xylan oligosaccharides and xylan 1-4-β-xylocidase to cleave the oligosaccharides into xylose monomers. Pérez and others (2002) have pointed out that four different enzymes are required to degrade the common hemicellulose O-acetyl-4-O-methylglucuronxylan: endo-1-4-β-xylanase, acetyl esterase, ∝-glucuronidase, and β-xylosidase.
The microbial degradation of pectic polysaccharides also is catalyzed by a group of specialized enzymes (pectinases) (Benen and others, 1996; Beldman and others, 1996; Bergmann and others, 1996; Hugouvieux-Cotte-Pattat and others, 1996). Pectinases may be divided according to their degradation mechanisms into polygalacturonases, pectin esterases, pectin lysases, and pectate lysases (Hoondal and others, 2002). As in the case of cellulose and hemicellulose, biological degradation systems ultimately depolymerize pectins to simple sugars.
The lignin polymers in vascular plants are composed of varying amounts of p-hydroxyphenylpropanoid, guaiacylpropanoid and syringylpropanoid monomeric units (Douglas, 1996; Saake and others, 1996). The most common linkages formed during lignin polymerization are β-O-4 ether linkages (fig. 1); however, Saake and others (1996) pointed out that other linkages such as ∝-O-4, β-β, β-5, and 5-5 also are present in lignins. Camarero and others (1994) have pointed out that, especially in grasses, the C-4 also may be attached to a hydroxyl group (C-4 phenol).
In woody terrestrial plants lignin is imbedded in the hemicellulose matrix of the secondary cell walls. Lignin deposition is one of the final stages of xylem-cell differentiation (Donaldson, 2001). The deposition takes place in the interlamellar voids of the cell walls. During the deposition, chemical bonds form between the lignin and hemicellulose carbohydrates (Sun and others, 2000). Lam and others (1992) have shown that cinnamic acid groups in wheat lignin are bound to polysaccharides by ester linkages. Two different mechanisms have been proposed for the lignin polymerization. Lewis and his co-workers (see Burlat and others, 2001) have proposed that lignin deposition is initiated at specific dirigent binding sites in the cell wall. Monolignol-derived free radicals polymerize at these sites. Burlat and others (2001) pointed out that it has been proposed that "* * *lignification occurs through an iterative process of lignin assembly (following primary chain formation) in a template guided manner." This process apparently favors β-O-4 linkages. Other workers, however, feel that lignin formation is a more "loosely ordered" process in which phenolic phenylpropanoid monomers other than p-hydroxyphenylpropanoid, guaiacylpropanoid, and syringylpropanoid monolignols can be incorporated into a lignin oligomer/polymer by undirected free-radical coupling (see Ralph and others, 2001). In either scenario the lignin oligomers ultimately link to the hemicellulose chains of the matrix.
Terashima and others (2002) have differentiated between lignin isolates and lignin as it exists in plant cells. They call the lignin that is chemically and physically bound to hemicellulose in plant cell walls protolignin. 13C NMR analysis indicates that protolignin is a heterogeneous polymer. For example, Terashima and others (2002) found that β-O-4 linkages are the most abundant bonds in wheat straw protolignin, but that β-5, β-1, and β-β linkages also are present. The protolignin forms a three-dimensional network in the hemicellulose matrix that apparently provides increased compressional strength to the aerial organs of land plants (Donaldson, 2001). The hemicellulose polymers are commonly linked to lignin polymers by benzyl ester, benzyl ether, and phenyl glycosidic linkages (Lawoko and others, 2003; Sun and others, 2000).
In as much as lignin occurs as hemicellulose-lignin complexes, lignin degradation cannot be considered independently of the hemicellulose degradation. For example, Wershaw and others (2003) isolated intact hemicellulose-lignin complexes from a natural black water derived from degradation of wheat straw. The complexes occur in the black water as soluble, high molecular weight molecules. The more hydrophilic hemicellulose chains probably constitute the exterior surfaces of the particles with the lignin chains in the interior portions of the particles. Therefore, degradation of the lignin will require prior degradation of the hemicellulose.
Lignin degradation by specialized microorganisms is relatively rapid. Ulmer and others (1983) found that lignin isolates were reduced to low molecular weight soluble products by the white-rot fungus Phanerochaete chrysosporium within 6 to 8 days. The enzymatic degradation of lignin by microorganisms generally involves both depolymerization and aromatic-ring cleavage (Crestini and others, 1998). This degradation of lignin is brought about by extra-cellular oxidative enzymes. The action of these enzymes generally has been studied using lignin model compounds and lignin isolates that have been mechanically and chemically freed from the surrounding hemicellulose matrix.
Crestini and others (1996) studied the degradation of lignin model compounds by the white-rot fungus Lentinus edodes. β–O-4 linkages were oxidized to arylglycerol compounds, and aromatic rings were cleaved. Cleavage of aromatic rings coupled with β–O-4 oxidation can lead to the formation of cyclic carbonate structures. The aromatic rings are cleaved by enzymatic systems that mostly follow the β–ketoadipate pathway. Harwood and Parales (1996) have reviewed the enzymatic reactions that take place in the β–ketoadipate pathway. A wide variety of aerobic soil bacteria and fungi secrete enzymes that convert aromatic compounds to aliphatic carboxylic acids. The first step of the conversion involves mono- or di-oxygenation of the aromatic ring to produce a dihydroxylated phenyl ring. Fission of the ring can take place either between two adjacent hydroxyl groups (ortho-fission) or adjacent to a hydroxyl group (meta-fission). Examples of these reactions are shown in figure 2. Schemes I and II (ortho-fission) in figure 2 lead to the production of β–ketoadipate. Scheme III is the first step of meta-fission, and Scheme IV is the first step in the gentisate pathway in which fission takes place between the carbon atom that is attached to a carboxylate group and an adjacent phenolic carbon. Schemes III and IV do not lead to the formation of β–ketoadipate (Fairley and others, 2002; Mohamed and others, 2001; Zaar and others, 2001).
In addition to the degradation of lignin polymeric units, the microbial degradation of other phenolic compounds, aromatic hydrocarbons, aminoaromatic compounds, and chlorinated aromatic compounds often follow the β–ketoadipate pathway in soils. Fused ring aromatic compounds also can undergo degradation by the β–ketoadipate pathway (Hinter and others, 2001). Similarly, carboxylate-substituted aromatic compounds other than lignin units may be degraded by the gentisate pathway (Fairley and others, 2002; Mohamed and others, 2001; Zaar and others, 2001).
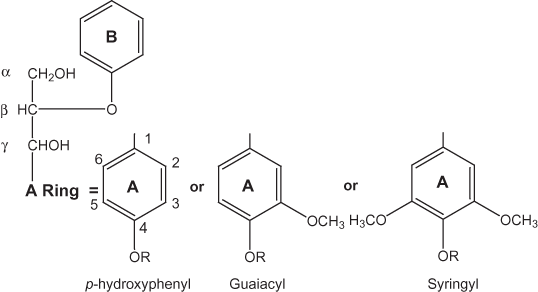
Figure 1. Lignin monomeric units.
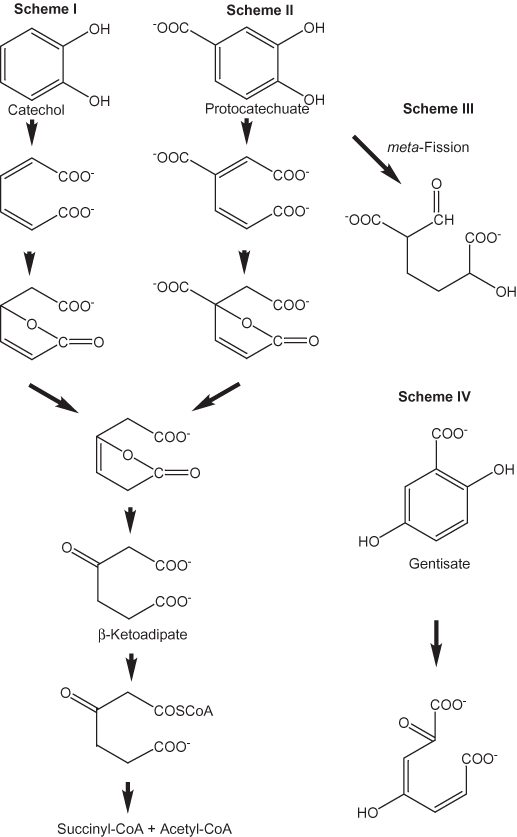
Figure 2. Lignin degradation pathways.
There are two types of polyester structural components in plants: cutin and suberin (Kolattukudy, 2001). The outer covering (cuticle) of the aerial parts of plants is composed of cutin. Suberin is a component of cork cells in the bark of vascular plants and in the epidermis, endodermis, exodermis, and phellem of roots; it also is deposited in wounds to seal them (Bernards, 2002).
Cutin is a heterogeneous polyester composed to a large extent of hydoxy-substituted, saturated C16 acids and unsaturated C18 acids (Kolattukudy, 2001). Some of the more common cutin-forming acids are shown in figure 3; a model of a cutin monomeric unit is given in figure 4. The cuticles of plants consist of the cutin polymer embedded in a matrix of soluble plant waxes.
Both bacteria and fungi secrete enzymes that degrade cutin. Cutin-degrading fungal enzymes have been studied more extensively than bacterial enzymes; however a bacterial cutinase has been isolated that hydrolyzes the ester linkages of cutin (Berto and others, 1999; Kolattukudy, 2001). The fungal enzymes have received more attention because of the damage caused by plant fungal diseases. Kolattukudy (2001) pointed out that a large number of phytopathogenic fungi secrete enzymes that hydrolyze cutin polyesters, thereby allowing the fungi to penetrate plant cuticles and harm the internal plant tissue. Cutin oligomers and monomeric fatty acids are produced during enzymatic cutin hydrolysis.
Whereas cutin is composed mainly of straight-chain fatty acids, suberin is made up of aromatic and aliphatic monomeric units. The suberin polymer appears to consist of polyaromatic and polyaliphatic domains (Bernards, 2002; Bernards and Lewis, 1998). Bernards and Lewis (1998) pointed out that "While it is clear that suberized tissues contain both polyaromatic and polyaliphatic domains, and that each of these has its own unique characteristics, whether there is a contiguous macromolecule that can be called suberin is an open question." They stated further that "* * *the notion of a heteropolymer, called suberin, must be questioned and it is perhaps more prudent to describe tissues as suberized, rather than as containing suberin per se."
Kolattukudy (2001) has reviewed the literature on the biodegradation of suberized tissue. He pointed out that suberized tissue is very resistant to degradation. Parameswaran and Wilhelm (1979) found in their study of the degradation of beech and spruce bark that suberized tissue was more resistant to degradation by soil microorganisms than other bark components. However, fungi can penetrate suberized tissue and degrade it (Kolattukudy and Espelie, 1989). This degradation leaves holes in the suberized tissue of bark and roots. Kolattukudy and Espelie (1989) pointed out that a number of fungal species produce enzymes that degrade suberized tissue. There are two different types of enzymes: those that degrade the aromatic domains and those that degrade the aliphatic domains. The esterase that catalyzed the hydrolysis of esterfied aliphatic acids of suberized tissue was shown to have identical properties to a cutinase produced by the same fungal species. All types of monomeric aliphatic acids were released from the suberized tissue by the enzyme. The enzyme or enzymes that degrade the aromatic domains have not been isolated. It appears that they mainly hydrolyze ester linkages; ether linkages appear to be resistant to attack.
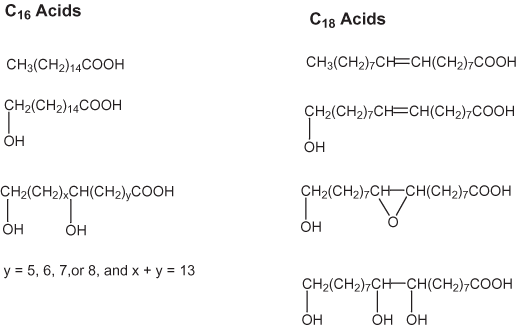
Figure 3. Cutin-forming acids.
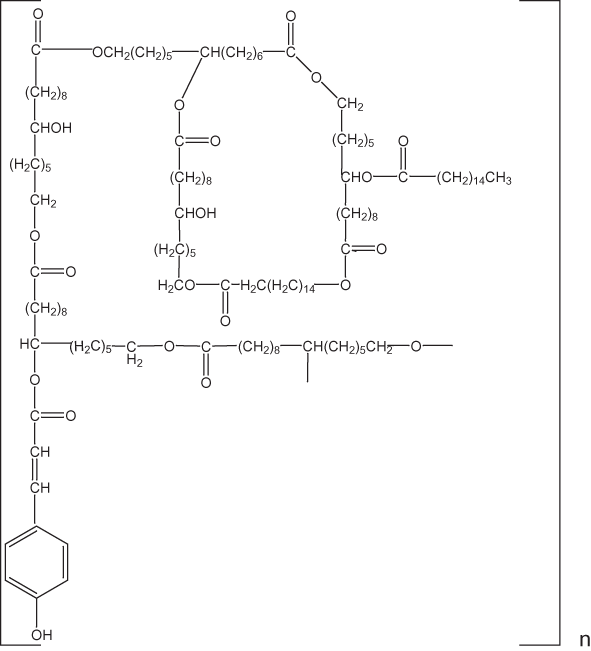
Figure 4. Model cutin monomeric unit according to Kolattukudy (2001).
Two groups of aliphatic acids will be covered in this section: (1) The low molecular weight acids and (2) the straight-chain fatty acids. Ohta (1989a) has reviewed the low molecular weight aliphatic acids. Some of these acids serve as metabolic intermediates in the citric acid and glyoxylate cycles of plants, and therefore, they are transient components of plant tissue. Their concentrations in a tissue will be dependent on the organs from which the tissue comes and on the time in the life cycle of the plant when a tissue sample is isolated. Examples of these acids are citric acid, succinic acid, fumaric acid, malic acid, pyruvic acid, and malonic acid. In addition to those acids that are constantly being cycled by plant metabolic processes, there are other low molecular weight acids that are end products of plant metabolism that generally accumulate in cell vacuoles. Examples of these acids are lactic acid, oxalic acid, and tartaric acid. These acids are commonly found in high concentrations in some plant species and in much lower concentrations in other species (see Ohta, 1989a, p. 267–268). Low molecular weight lactones and other alicyclic compounds also are found in some plant species (Ohta, 1989b).
The fatty acids accumulate in plant tissue as energy reserves. They commonly occur as triglyceride esters; however, free fatty acids are also found in plant tissue (Zinkel, 1989). Fatty acids are readily degraded by soil microorganisms. The degradation pathways of fatty acids are similar in all organisms and will not be described in detail here. In general, fatty acids undergo β-oxidation in which two carbon atoms are successively removed from the ends of the chains until the last two carbon atoms are left as acetyl-CoA. The acetyl residue then undergoes oxidation to carbon dioxide. Fatty acids also can undergo ω-oxidation in which a terminal methyl group is oxidized by monoxygenase enzymes to a carboxylate group, thereby yielding an ∝,ω-dicarboxylic acid (Eschenfeldt and others, 2003).
Kolattukudy and Espelie (1989) have reviewed the plant waxes that are associated with cutin and suberin. The most common components of plant waxes are long-chain alkanes, long-chain primary and secondary alcohols, fatty aldehydes and ketones, fatty acids, and monoesters of fatty acids and long-chain alcohols. So-called very-long-chain fatty acids (greater than 18 carbon atoms in length) are the precursors of most components of waxes (Mariani and Wolters-Arts, 2000). The relative proportions of the wax compounds vary from plant species to plant species and from one type of organ to another. Plant waxes are present both as layers covering plant cuticles and imbedded in the cutin of the cuticles (Heredia, 2003).
Plant waxes are relatively resistant to microbial degradation; however, there are some microorganisms that produce enzymes that can degrade them. Some groups of microorganisms, such as those belonging to the genus Acinetobacter, produce wax esters for energy storage (Ishige and others, 2003; Rontani and others, 2003). These microorganisms, therefore, must possess enzyme systems that can degrade wax components. Berto and others (1999) have shown that the spores of the phytophathogenic fungus Alternaria brassicicola contain a lipase that can hydrolyze cuticle waxes. McKenna and others (2002) have isolated strains of non-streptomycete actinomycetes from soils that produce enzyme systems that can degrade plant waxes. They have used these organisms to ameliorate water repellency in a soil.
Proteins are the most abundant polymeric components in living cells. With the exception of some bacterial peptides and some peptide antibiotics, all proteins are composed of the same set of twenty L-amino acids. The amino acids of peptides and proteins are linked together by peptide bonds formed by the condensation of the ∝-carboxyl group of one amino acid with the ∝-amino group of another amino acid. Because peptide bonds are formed by the elimination of water, they are susceptible to hydrolysis; however, the bonds are stable because their activation energies of hydrolysis are high. Hydrolysis of peptide bonds requires strongly acidic or basic conditions or enzymatic catalysis.
The enzymes that hydrolyze peptide bonds are called proteases. Soil microorganisms secrete proteases to hydrolyze proteins into their constituent amino acids (Martens and Loeffelmann, 2003). The amino acids so produced then are utilized by the microorganisms as sources of carbon and nitrogen (Martens, 2001, Reitzer, 1996a,b; McFall and Newman, 1996). Microorganisms can utilize the amino acids in two possible ways. They can reduce them to ammonia (or ammonium ion) and hydrocarbon compounds and then use the ammonia to produce new amino acids and aminosugars for incorporation into larger polymers, or they can assimilate the existing amino acids and use them as building blocks for proteins and other polymeric species (Reitzer, 1996b). Assimilation of amino acids can involve partial degradation; for example, members of Enterobacteriaceae convert L-arginine, L-glutamine, L-histidine, and L-proline to L-glutamate (McFall and Newman, 1996). The L-glutamate can be utilized as an energy source via the ∝-ketoglutamate pathway while still maintaining much of the carbon skeleton and the C-N bond intact.
Microbial biomass also contributes to NOM. The chemical composition of microorganisms is much different from that of plants; and therefore, the composition of the soil microbial biomass will be different from that of plant-derived soil organic matter (SOM). For example, bacteria are composed of proteins, RNA and DNA, lipids, lipopolysaccharides, peptidoglycans, glycogen, polyamines, various metabolites, cofactors, and inorganic ions (Neidhardt and Umbarger, 1996). Because microbial biomass constitutes only about 1 to 5 percent of the total carbon in most soils (Angers and others, 1993; Anderson and Domsch, 1993; Jenkinson and Ladd, 1981) one might not expect it to greatly alter the composition of SOM. However, the presence of certain constituents, such as N-acetylglucosamine and N-acetylgalactosamine, are diagnostic of microorganisms. In contrast to soils, microbial biomass can be a major component of dissolved and suspended NOM in natural waters.
Tannins are commonly divided into two groups: hydrolysable and nonhydrolysable tannins. The hydrolysable tannins contain gallic acid moieties (fig. 5). Gallic acid apparently is produced in plants by degradation reactions (Gross, 1999); Haslam (1998) concluded that gallic acid is formed mainly from the dehydrogenation of 3-dehydroshikimate. It then apparently undergoes esterification to form β-1,2,3,4,6-pentagalloyl-D-glucose, which appears to be an important precursor in the formation of the majority of gallotannins and ellagitannins in plant tissue. Ellagitannins are formed by oxidative coupling that results in the formation of C–C bonds between adjacent galloyl groups as shown in figure 6 (Gross, 1999). Hydroxyl groups adjacent to acid groups in these dimers can undergo esterification to form internal esters (lactones), as shown in figure 6. The dimer shown in figure 6 can undergo further polymerization to form larger oligomers. The esterification of two gallic molecules produces a compound called a depside.
The nonhydrolysable or condensed tannins are oligomers of phenolic flavan-3-ol (catechin) units as shown in figure 7 (Haslam, 1998). The biosynthesis of condensed tannins starts with the conversion of L-phenylalanine to cinnamate; further steps in the biosynthesis are outlined by Haslam (1998). As shown in figure 7, the flavan-3-ol units generally are linked to each other at the 4 and 8 positions to form proanthocyanidin oligomers and polymers; however, other linkages such as 4–6 also exist (see Ferreira and others, 1999). Oligomers of 2 to 6 units generally are soluble; larger oligomers are insoluble; the monomeric units of these oligomers are called anthocyanidins. Anthocyanidins often exist as glycosides in plants; these glycosides are called anthocyanins. The polymeric species are called proanthocyanins. The larger insoluble oligomers and polymers are generally in higher concentration in plant tissue than the soluble oligomers. Flavan-3-ol units also can be linked to gallotannins or ellagitannins to form "complex tannins" (Ferreira and others, 1999).
It has been shown that hydrolysable tannins are degraded by some filamentous fungi. These fungi produce enzymes called tannases that catalyze the hydrolysis of ester bonds between phenolic acid moieties and alcohols or polyols (Scalbert, 1991; Gross, 1999). Depside bonds also are cleaved by tannases. Some fungi also secrete polyphenoloxidases that appear to oxidize tannins. The biodegradation of nonhydrolysable tannins has been much less well studied than that of the hydrolysable tannins; however, it appears that some yeasts can depolymerize them (Scalbert, 1991).
Tannins exhibit antimicrobial activity towards many types of fungi, bacteria, and yeasts. Scalbert (1991) has reviewed the antimicrobial properties of tannins. Four different mechanisms have been proposed to explain these properties: (1) inhibition of extracellular microbial enzymes, (2) deprivation of substrates required for microbial growth, (3) inhibition of oxidative phosphorylation in the metabolic processes of the organisms, and (4) deprivation of iron and other metals to the organisms by metal binding. The inhibition of some enzymes by tannins may be due to binding of the enzymes to the tannins. It is well known that tannins precipitate proteins and polysaccharides by forming complexes with them.
Terpenes are organic compounds that are composed of isoprene units arranged either in open chains or cyclic structures. They generally exist as monoterpenes (two isoprene units), sesquiterpenes (three isoprene units), diterpenes (four isoprene units), triterpenes (six isoprene units), or tetraterpenes (eight isoprene units). Wagner and Elmadfa (2003) pointed out that most naturally occurring terpenes are cyclic unsaturated hydrocarbons with attached oxygen-containing functional groups. At least 15,000 terpenes have been identified. Wagner and Elmadfa (2003) stated that "In nature terpenoid molecules are implicated in almost every interaction between plant and animal, plant and plant, or plant and microorganisms as phytoalexins, insect antifeedants, defense agents, pheromones or signal molecules." Consumption of terpenes in fresh fruits and vegetables is purported to have important health benefits.
Isoprene, the simplest terpene, constitutes about 50 percent of the biogenic non-methane hydrocarbons in the atmosphere (Claeys and others, 2004). Isoprene and some of the monoterpenes constitute the most abundant group of reactive biogenic compounds in the atmosphere (Limbeck and others, 2003; Simpson and others, 1999; Guenther and others, 1995). Limbeck and others (2003) have shown that "humic-like substances" form when isoprene reacts with sulfuric acid in aerosols. These substances will precipitate from the atmosphere and contribute to the NOM pool.
Resin acids from trees constitute a major source of terpenes in the environment (Yu and others, 2000). Resin acids are composed of tricyclic diterpenoid carboxylic acids. Yu and others (2000) isolated a number of bacterial strains from hydrocarbon-contaminated Arctic tundra soils that degrade resin acids.
Leenheer and others (2003) have identified degradation products of terpenes in landfill leachates, and surface and ground waters. They pointed out that alicyclic terpenoid structures are more resistant to bacterial degradation than are open-chained or alkene terpenoid structures. In addition, structures containing aliphatic quaternary carbons and branched methyl groups are resistant to microbial degradation. Leenheer and others (2003) have shown that bands characteristic of quaternary aliphatic carbon atoms and branched methyl groups in the 13C NMR spectra of NOM isolates are indicative of the presence of resistant terpenoid structures in the isolates.
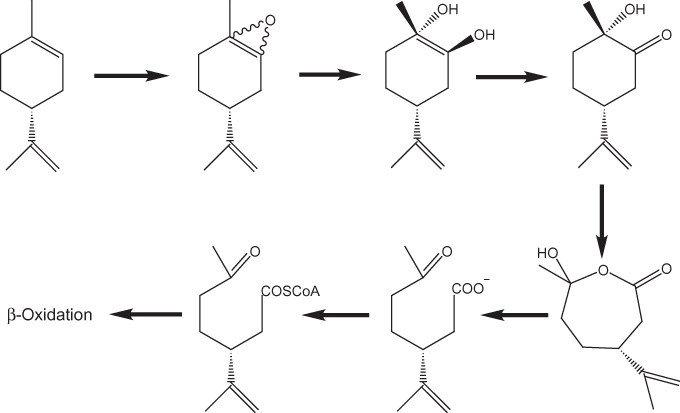
Some organisms, however, produce enzymes that degrade cyclic terpenes. Van der Werf and others (1999) found that ring scission is one of the pathways of limonene degradation brought about by a strain of the gram-positive bacterium Rhodococcus erythropolis isolated from a ditch sediment. Limonene is a very abundant monocyclic monoterpene that is found in more than 300 plant species. Other studies (see references cited in van der Werf and others, 1999) also have shown that enzymatically catalyzed β-oxidation of the alicyclic ring of limonene takes place. Other oxidation products also are produced during microbial degradation of limonene. The pathway for Rhodococcus erythropolis degradation of (+)-(4R)-limonene according to van der Werf and others (1999) is shown in figure 8.
Harder and Probian (1995) found that anaerobic bacterial cultures can oxidize some monoterpenes in the presence of nitrate as an electron acceptor. Acyclic, monocyclic, and bicyclic monoterpenes were degraded. Resin acids also undergo anaerobic degradation (Martin and others, 1999). Molecules of resin acids generally are composed mainly of hydrophobic groups, and they readily sorb to suspended mineral particles. When the mineral particles settle out of suspension to form sediments, the sorbed resin acids are transported to an anoxic environment. The anaerobic biodegradation of the resin acids is poorly understood; however, it appears that some resin acids undergo aromatization and decarboxylation to form alkylated polycyclic aromatic hydrocarbons (Martin and others, 1999). Complete mineralization of resin acids by nitrate reducing bacteria in anaerobic environments also may take place.
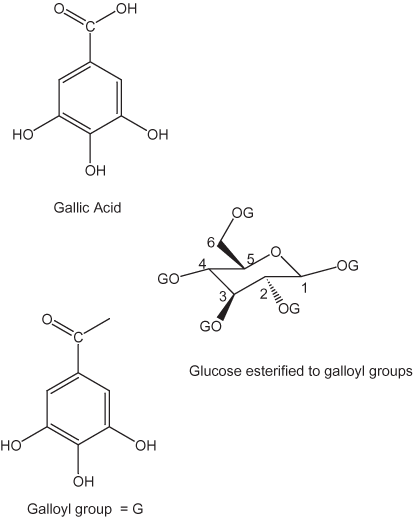
Figure 5. Hydrolysable tannin units.
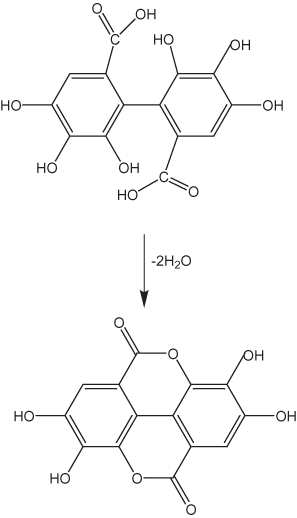
Figure 6. Ellagitannin formation.
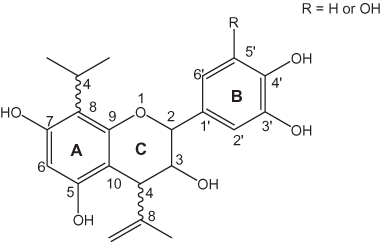
Figure 7. Nonhydrolysable tannins.
Pyrolytic reactions lead to the formation of the so-called black carbon (BC) fraction in soils. BC is the least studied component of soil organic carbon (SOC). BC has been detected in many soils and sediments (Cambardella and Elliot, 1992; Karapanagiotti and others, 2000; Schmidt and others, 1999; Skjemstad and others, 1996; Skjemstad and others, 2002) and is probably present in most soils. Gélinas and others (2001) estimated that BC accounts for as much as 55 percent of SOC. They divided BC into two components: char/charcoal BC (CBC) and soot/graphitic BC (GBC). The CBC fraction is formed by the charring biomass mainly of plant origin, whereas the GBC fraction is formed by vapor-phase reactions. Gélinas and others (2001) pointed out that GBC particles are much smaller than CBC particles, and that they are "* * *rapidly dispersed through aerosol formation and atmospheric transport and deposition." The CBC particles, on the other hand, generally will remain in the soil near where they are produced. The GBC particles may be transferred long distances before they precipitate out of the atmosphere and become incorporated into soils and natural waters.
Simoneit (2002) has reviewed the literature on the organic compounds that are associated with GBC particles produced by the burning of wood and other biomass. Simoneit (2002) pointed out that during the heating of wood, the sequence of decomposition with increasing temperature is hydrolysis, oxidation, dehydration, and finally pyrolysis. During the heating process the constituents of wood are converted into combustible volatile compounds, tars, and char. When the ignition temperature of the volatiles or the tars is reached, spontaneous exothermic oxidation (combustion) begins. During combustion, resins, the thermal decomposition products of cellulose, hemicellulose, and lignin, and water are volatilized. These vapors then undergo either partial or complete combustion. Some of the products of partial combustion are deposited on soot particles.
The pyrolysis of cellulose, the most abundant constituent biomass, has been studied more extensively than that of any other biomass constituent. Shafizadeh (1982, 1984) has delineated two different pathways for cellulose pyrolysis. At temperatures below about 300°C bond scission, formation of free radicals, dehydration, and formation of carbonyl, carboxyl, and hydroperoxide groups predominate. These reactions are followed by production of carbon monoxide, carbon dioxide, and char. The carbon monoxide is apparently the result of decarbonylation and the carbon dioxide of decarboxylation. At temperatures above 300°C bond cleavage by transglycosylation, bond fission, and disproportionation reactions take place. This second pathway (fig. 9) leads to the formation of anhydro sugars such as levoglucosan by dehydration. (See Nimlos and others, 2003, for a detailed discussion of dehydration of carbohydrates.) Sanders and others (2003) have developed a more detailed model of the saccharides. They found that at temperatures between 300 and 600°C low-molecular weight oxygenated compounds such as anhydro sugars and simple aromatic compounds are formed. At higher temperatures polynuclear aromatic hydrocarbons are produced. However, McGrath and others (2003) found that polynuclear aromatic hydrocarbons form between 300 and 650°C. Polynuclear aromatic hydrocarbons also are formed from pyrolysis of lignin (Sharma and Hajaligol, 2003) and amino acids (Sharma and others, 2003).
Shafizadeh (1984) pointed out: "The production of volatiles leaves a solid residue that is neither intact substrate nor pure carbon, but a different material at various stages of charring and carbonization. The intermediate chars are characterized by the functional groups present (including aromatic and olefinic structures); a high concentration of free spins trapped in a rigid structure or stabilized by aromatic and olefinic structures; a large surface area; and high degree of reactivity." Fused ring aromatic structures develop during the charring process.
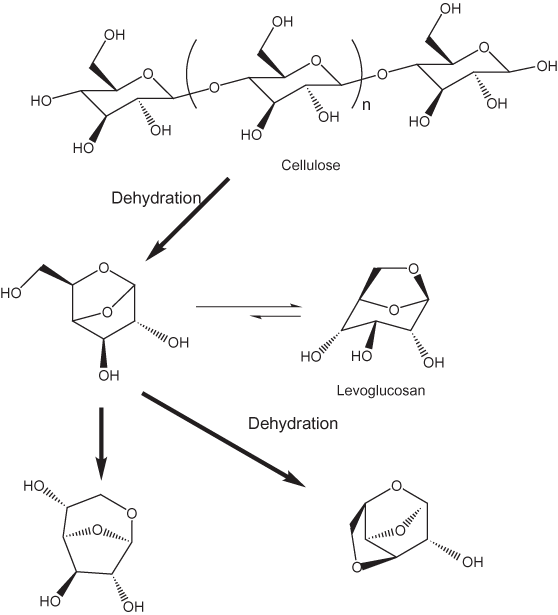
Figure 9. Formation of anhydro sugars.
Sharma and others (2003) studied the pyrolysis of ∝-amino acids in an inert atmosphere. They identified polycyclic aromatic hydrocarbons and nitrogen-containing polycyclic aromatic compounds as products. At low temperatures (300°C) single-ring heterocyclic compounds also were formed. Further research will be necessary to determine the applicability of these results to pyrolysis in air.
Simoneit (2002) pointed out that "Lignin pyrolysis products are major components in the fine aerosol fraction of smoke samples from fires burning wood* * *." Rogge and others (1998) analyzed the smoke from burning pine and oak wood in residential fireplaces. They identified approximately 200 different organic compounds including n-alkanes, cyclohexylalkanes, n-alkanals, n-alkanoic acids, alkenoic acids, dicarboxylic acids, resin acids, hydroxylated and methoxylated phenols, lignans, substituted benzenes and benzaldehydes, phytosterols, polycyclic aromatic hydrocarbons, and oxy-polycyclic aromatic hydrocarbons. They attributed the hydroxylated and methoxylated phenols and the substituted benzenes and benzaldehydes to pyrolysis of lignin. The lignin-derived compounds were the most abundant products in the wood smoke. Resin acids (terpenes) also were abundant in the pine smoke.
Terpenes undergo dehydrogenation reactions to form products such as dehydroabietal and dehydroabietic acid during burning of biomass (Simoneit, 2002). Further heating of diterpenes with the abietane skeleton leads to decarboxylation to form dehydroabietin, and finally to aromatization to form retene (Oros and others, 2002). Simoneit and others (1993) found that dehydroabietic acid is the dominant compound in pine smoke. Many terpenes are volatile and are driven off during burning along with water vapor. Depending on the temperature of the fire, some or all of the terpene vapors will completely burn. If the fire temperature is not too high, some of the terpenes will remain intact and be transported along with other combustion products (Rogge and others, 1998).
Oros and others (2002) have analyzed dichloromethane/methanol-soluble fractions from soils and litters from forests, grasslands, and chaparral landscapes before and after burning. They identified a number of diterpenes and triterpenes in the dichloromethane/methanol extracts of some of their samples. In soils from some of the burned areas, they also detected combustion products such as dehydroabietic acid and retene.
Oros and others (2002) detected n-alkanes, n-alkanoic acids, n-alkanols, and phytosterols in all of the extracts they analyzed. The first three of these groups of compounds are components of epicuticular plant waxes. The phytosterols are components of plant lipid membranes and plant waxes. Biomarkers for specific plant species or types of plants are found within all of these categories of compounds.
Oros and others (2002) detected specific alterations in all of the categories of compounds brought about by fire. They pointed out, for example, that the n-alkanes of the epicuticular waxes of Rye grass are composed of odd numbers of carbon atoms from 25 to 33, with a strong odd carbon number predominance. In unburned soil covered by Rye grass, the range of carbon atoms was from 15 to 31 with an odd carbon number predominance. After burning, the n-alkanes of the grassland soil had a range of carbon atoms from 15 to 33 and an even stronger odd carbon number predominance than in the unburned soil. However, the fatty acids in the unburned grassland soils had a strong even carbon number predominance with carbon numbers between 16 and 24. After burning the carbon numbers ranged between 14 and 32, and even carbon numbers were still predominant, although to a lesser degree than in the unburned soil.
Photochemical reactions are classified as being either direct or indirect. Direct reactions are those in which chromophores in the molecule being degraded absorb electromagnetic radiation (light) that promotes the degradation. Chromophores can absorb radiation in the visible or ultraviolet range, or both. The added electromagnetic energy causes a chromophoric group in a molecule to go from its ground state to an excited state. The lower wavelength limit for solar radiation reaching the surface of the earth is approximately 300 nanometers (nm), and therefore, only chromophores that absorb radiation of 300 nm or greater will undergo transitions to excited states. Aromatic and heteroaromatic functional groups, and conjugated polyenes and carbonyl groups undergo π-π* and n-π* transitions at these wavelengths; most other functional groups commonly present in NOM absorb at wavelengths shorter than 300 nm and are, therefore, inactive (Brown, 1999). The energy changes for these transitions can be of the order of the energy involved in forming a covalent chemical bond (Pine and others, 1980). The work of Osburn and others (2001), who studied the photodegradation of DOM from bog water, provides an example of direct degradation. They found that aromatic functional groups were preferentially degraded by solar radiation. Aliphatic functional groups that do not absorb solar ultraviolet radiation were more stable than the aromatic groups when exposed to solar radiation.
NOM also may undergo photochemical reactions by indirect processes in which photo-activated organic or inorganic species react with unactivated molecules (Goldstone and others, 2002; Faust, 1999; Larson and Marley, 1999; Zafiriou and others, 1984). Zafiriou and others (1984) pointed out that there is evidence that manganese, iron, copper, mercury, tantalum, carbon, nitrogen, oxygen, sulfur, chlorine, bromine, and iodine species enter into photochemical reactions in surface waters. Thus, for example, photoactivated oxygen-containing species could react with NOM.
A brief overview of direct and indirection photochemical reactions that take place in surface waters will be given in this section. In addition, the contribution to the NOM pool of chemical compounds produced by solar irradiation of plant components in the atmosphere will be discussed. A further complication arises because photodegradation often takes place in concert with biodegradation. Zepp and others (1995) pointed out that biodegradation may be accelerated or retarded by increases in solar ultraviolet radiation depending on the properties of the particular system undergoing solar irradiation.
Aromatic functional groups are plentiful in NOM. Richard and Grabner (1999) state that hydroquinone is particularly relevant environmentally because it absorbs radiation in the ultraviolet portion of the solar spectrum, and it has long-lived triplet states that are efficiently populated. Joschek and Miller (1966) found that irradiation of hydroquinone both in the presence and absence of oxygen in water produced similar products. Oxidation and hydroxylation products were formed along with a C–C dimer, 2,2',6,6'-tetrahydroxybiphenyl. They proposed the reaction mechanisms shown in figure 10 (Scheme I) to explain the products they detected. Richard and Grabner (1999) proposed a slightly different mechanism for the formation of the tetrahydroxybiphenyl product in which a triplet-state hydroquinone intermediate reacts with p-benzoquinone that also is produced during the photolysis as shown in figure 10 (Scheme II). Polycyclic aromatic hydrocarbons undergo similar types of direct photochemical reactions in water (Pagni and Sigman, 1999).
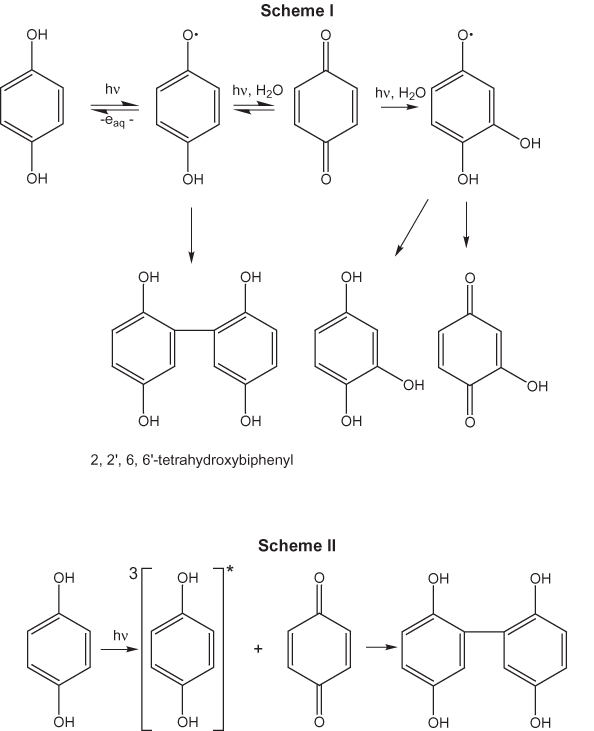
Indirect photochemical reactions are brought about by both organic and inorganic species. The most abundant group of organic compounds in natural waters is DOM. The colored constituents of DOM (humic substances) in natural waters participate in indirect photochemical reactions (Zafiriou and others, 1984). Although all the chromophores in DOM have not been well characterized, it is likely that they are mainly aromatic moieties. Zepp and Schlotzhauer (1983) have shown that the presence of algal material in fresh water accelerates the photodecomposition of nonionic organic compounds. The authors found in their study of the photo-degradation of aniline, m-toluidine, p-chloroaniline, parathion, and methyl parathion that these compounds were reversibly sorbed by the algal cells and that the degradation took place on the surfaces of the cells. The photosensitizing agents on the cell surfaces were not identified.
Photolysis of DOM produces hydroxyl radicals (OH•). Vaughan and Blough (1998) photolyzed Suwannee River fulvic acid (SRFA), both aerobically and anaerobically, in water. They found that under anaerobic conditions hydroxyl radicals were produced in proportion to the amount of SRFA present. They concluded from their observations that the OH• radicals were produced by direct photolysis of SRFA "* * *through a dioxygen-independent pathway* * *". They proposed that a possible mechanism for production of OH• radicals in the absence of dioxygen is through the abstraction of hydrogen from water by photo-activated (triplet state) benzoquinone groups in the SRFA.
Goldstone and others (2002) showed that OH• radicals react with SRFA to produce low-molecular weight organic acids (for example, formic, acetic, malonic, and oxalic acids). The photo-bleaching rates (reaction rates) they observed for SRFA were relatively slow, and they concluded that the OH• radical reaction with DOM only is able to account for a small part of observed photo-bleaching rates in natural water systems. The only exceptions would be in waters with high iron concentrations where the photo-Fenton reaction (Fe(II) + H2O2 → Fe(III) + OH• + OH-) would produce large concentrations of OH•. Brinkmann and others (2003) irradiated a bog-lake water with simulated solar ultraviolet radiation. They proposed that the observed production of formic, acetic, pyruvic, oxalic, malonic, and succinic acids was the result of a combination of photochemical and biochemical degradation reactions.
In addition to Fe(II), many other inorganic species produce reactive free radicals when irradiated with solar ultraviolet radiation in water (Faust, 1999). The most important of these are dioxygen (O2), nitrate ions, and Fe(III) and Cu(II) ions. Inorganic surfaces also catalyze the photodecomposition of organic compounds (Ollis, 1985; Ollis and others, 1991; Pagni and Sigman, 1999). Solar illumination of an oxide semiconductor surface produces excited electrons (e-) and holes (h+); thus, the excited electrons are reducing agents and the holes are oxidants. Concerning the excited electrons and the holes, Ollis and others (1991) pointed out "These can migrate to the oxide surface and participate in half-cell reactions that are part of a closed, catalytic cycle." In natural waters the photodecomposition of organic compounds, no doubt, is brought about by a combination of reactions, some of which take place in solution and some on surfaces.
Precipitation of organic compounds produced by photo-oxidation in atmospheric aerosols also can contribute to DOM. Isoprene is the most abundant biogenic nonmethane hydrocarbon in the atmosphere. Claeys and others (2004) identified two diastereoisomers of 2-methyltetrol in natural aerosols from the Amazonian rain forest. They attributed these compounds to photo-oxidation of isoprene by OH• radicals in the aerosols.
Free radical oxidative degradation processes take place during the later stages of leaf senescence. Leaf senescence is one of the major processes leading to the formation of DOM. In living plant tissue the reactions between the chemical components in plant cells are tightly controlled. The early stages of senescence also are highly regulated with specialized metabolic pathways being activated (Dangl and others, 2000). The enzyme systems active in these metabolic pathways degrade plant polymers to monomeric units that are then transported to storage organs for reuse during the next growing season. For example, carbohydrates and proteins are depolymerized to monosaccharides and amino acids respectively. Phospholipid cell-membrane components are degraded to saturated and unsaturated fatty acids. Nucleic acids and plant pigments also are degraded.
During the later phases of senescence, death of the leaf takes place, and the various compartments in the plant cells rupture and their contents mix together. After the cellular and subcellular walls rupture, chemical species produced in one subcellular compartment will be exposed to all of the chemical components in the leaf tissue. Oxygen-containing free radical species such as superoxide, hydroxyl, peroxyl, alkoxyl, polyunsaturated fatty acid, and semiquinone radicals are abundant in the cytoplasmic mixtures formed as cell walls rupture (Leshem, 1988; Philosoph-Hadas and others, 1994). These free radical species depolymerize plant biopolymers that have survived the earlier enzymatically-mediated depolymerization reactions. The amino acids, peptide, simple sugars, fragments of polyphenolic polymers, and other biopolymers produced are then leached from the dead leaves to become components of DOM (Wershaw and others, 1998a,b).
In addition to depolymerization reactions, plant phenolic compounds such as hydrolysable and nonhydrolysable tannins can undergo oxidative coupling reactions. The most extensively studied sequence of coupling reactions is that which takes place during tea fermentation. The processing of tea is initiated by withering and mechanically macerating the tea shoots to rupture the cell walls; this is followed by fermentation in a high-humidity environment where oxidation reactions catalyzed by polyphenol oxidases and peroxidases take place (Baruah and Mahanta, 2003). Haslam (2003) has reviewed the reactions that take place during tea fermentation. Two major groups of phenolic compounds have been identified in fully fermented tea (black tea): theaflavins and thearubigins. Theaflavins constitute as much as 6 percent and thearubigins as much as 75 percent of the solids in brewed tea. Theaflavins are characterized by the presence of the seven-carbon atom benzotropolone ring (B´ in fig. 11). Much less is known about the structures of thearubigins.
Haslam (1998) has pointed out that the benzotropolone ring arises from oxidative condensation of the otho-dihydroxyphenyl B ring of an epicatechin with the otho-trihydroxyphenyl B ring of an epigallocatechin (fig. 11). In the mechanism given by Haslam (1998), two of the three hydroxyl groups of the epigallocatechin B ring are oxidized to carbonyl groups before ring opening takes place. Davies and others (1999) show a slightly different mechanism in which all three of the hydroxyls are oxidized to carbonyls prior to ring opening.
Haslam (2003) speculated that thearubigins have structures similar to those that have been proposed for the products of the auto-oxidation of pyrogallol in basic solutions (fig. 12). The pyrogallol products result from nonezymatically catalyzed oxidative coupling reactions. A sequence of possible oxidation reactions for thearubigins proposed by Haslam (2003) is shown in figure 13. Davies and others (1999) suggested that thearubigins are composed of units such as those shown in figure 14. All of these proposed structures are highly speculative; at the present time there are no definitive data on thearubigin structures.
Coupling reactions of phenolic compounds also also have been extensively studied in the aging of wines (Salas and others, 2003). The reactions in red wines have been of particular interest because the red pigments are flavonol glycosides (anthocyanin pigments shown in fig. 15). Proanthocyanidins (fig. 16), condensed oligomers of two to six anthocyanidin (aglycone) units linked through the C-4 and C-8, also are present in red wines. The proanthocyanidins undergo cleavage of the 4–8 bond forming flavanols in the process. These flavanols, in turn, react with anthocyanin flavylium cations to form a colorless flavene dimer. The dimer, in turn, can undergo oxidation to the red flavylium cation (fig. 15) or conversion to a xanthylium salt or a bicyclic structure (fig. 17).
The flavylium cation is the dominant form for anthocyanins in the pH range between 1 and 3 (Kähkönen and Heinonen, 2003); at higher pH values the quinonoidal anhydrobases and anionic quinonoidal anhydrobases form as shown in figure 18 (Haslam, 1998). In aqueous solution flavylium cations are converted to carbinol bases with time or dilution at pH values between 4 and 5 (Jurd, 1963; Kähkönen and Heinonen, 2003).
The oxidative reactions outlined above illustrate that the types of abiotic reactions that the polyphenolic components of NOM can undergo. It is important to point out, however, that enzymatic systems also are capable of catalyzing many of these reactions, and therefore, in environmental systems it often is not possible to distinguish between the results of biotic and abiotic oxidative mechanisms.
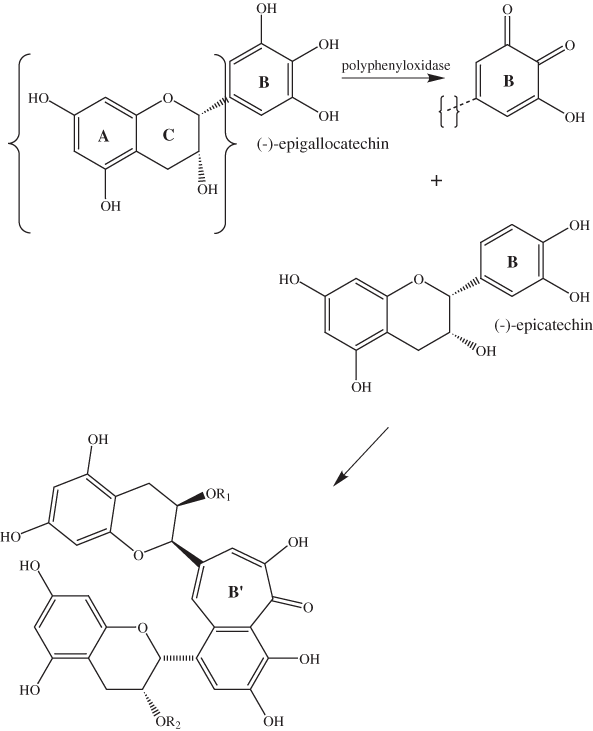
Figure 11. Formation of benzotropolone rings according to Haslam (1998).
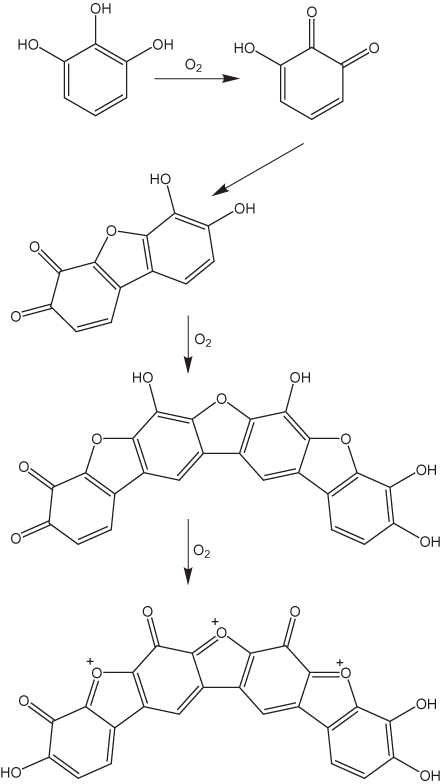
Figure 12. Auto-oxidation of pyrogallol (Haslam, 2003).
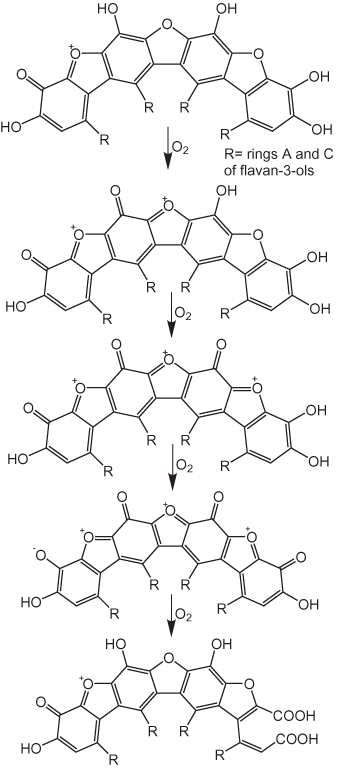
Figure 13. Possible oxidation reactions of thearubigins according to Haslam (2003).
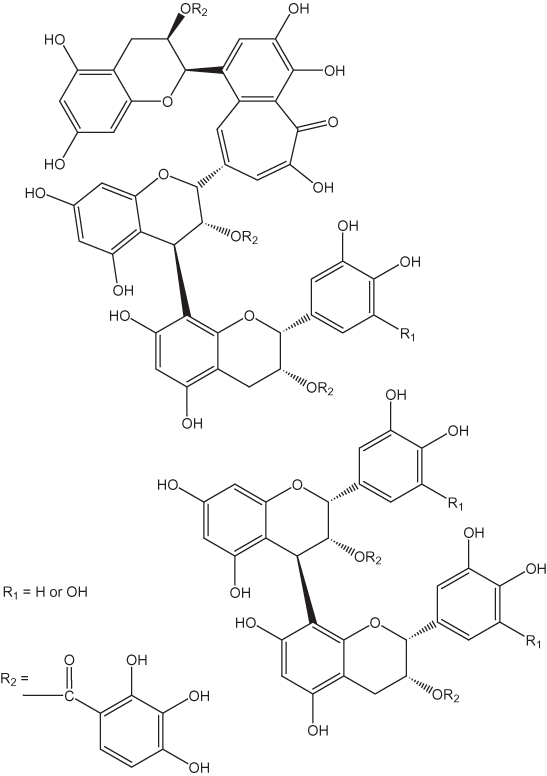
Figure 14. Thearubigin units according to Davies and others (1999).
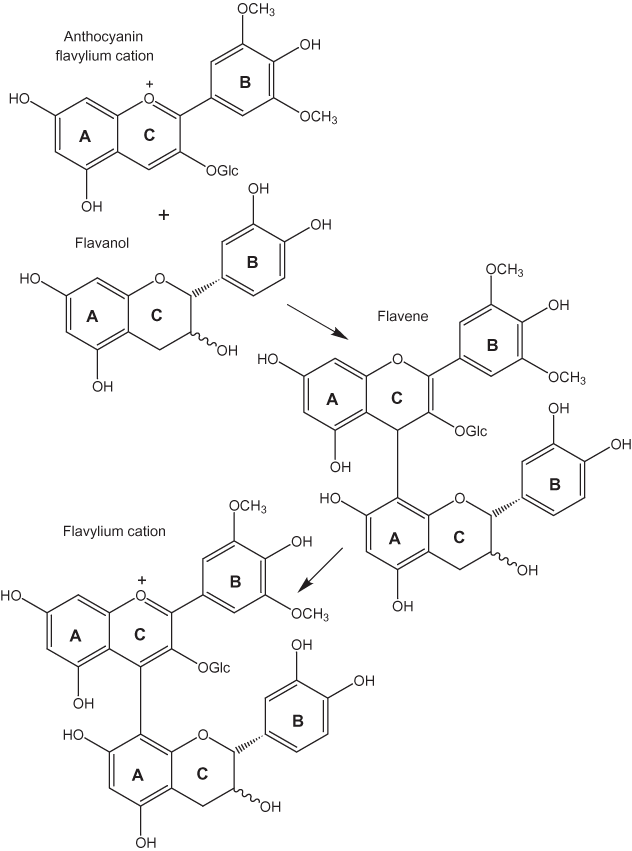
Figure 15. Formation of anthocyanin pigments (Salas and others, 2003).
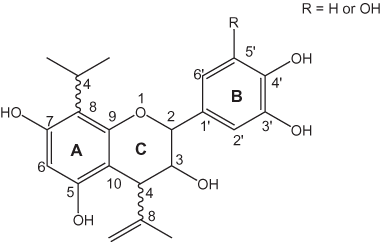
Figure 16. Proanthocyanidin oligomers.
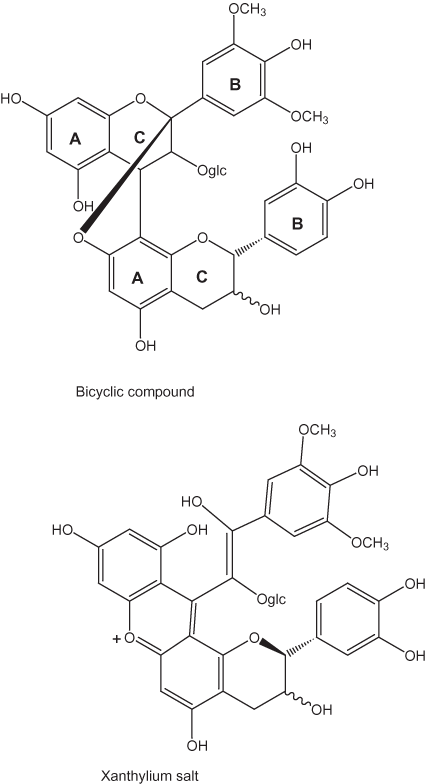
Figure 17. Bicyclic and xythylium salt forms of flavene dimers.
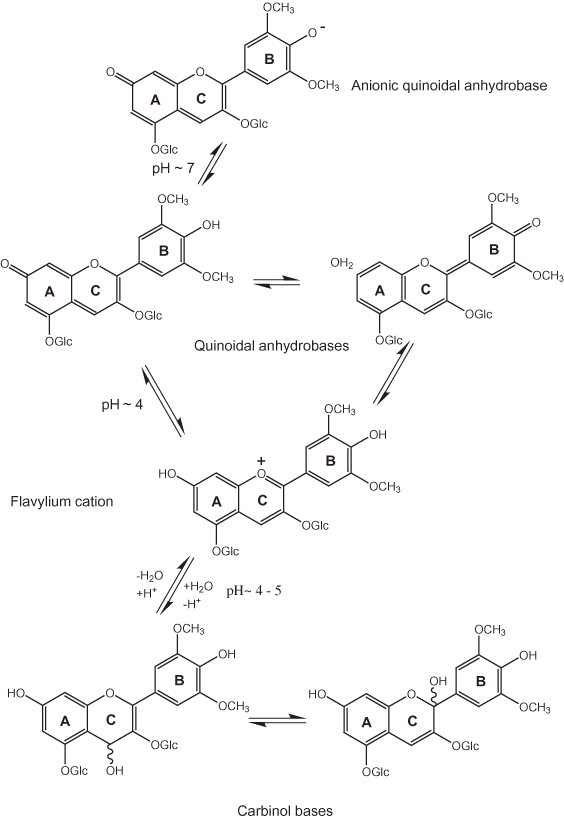
Figure 18. Effect of pH on the structure anthocyanins.
Another possible non-enzymatic reaction pathway is the browning reaction that commonly takes place in foods. In this type of reaction, reducing sugars react with free amino groups in compounds such as amino acids and proteins to form the so-called Maillard products. Amino acids may have only a single ∝ amino group or more than one amino group as in the case of lycine with an additional ε amino group. Both primary and secondary amines are susceptible to reaction with sugars (Friedman, 1996; Ledl and Schleicher, 1990). The first step in the reaction is the formation of a Schiff base followed by re-arrangement to an Amadori product (figure 19). Further reaction leads to the formation of heterocyclic nitrogen compounds (see Friedman, 1996; Ledl and Schleicher, 1990). The distribution of reaction products will be dependent on relative rates of the different steps in the reaction sequence. Unfortunately, the kinetics of the different reaction steps are complex and much work remains before definitive predictions on relative concentrations of reaction products can be made (van Boekel, 2001).
Browning reaction products are most commonly formed during the cooking of foods. Many of these products are important in improving the taste, aroma, and appearance of cooked foods. However, mutagenic, carcinogenic, and other types of harmful browning products also can be formed during the cooking process (Friedman, 1996). In addition, Tauer and others (2004) found that increased amounts of Maillard products inhibited enzymatic fermentation of carbohydrate-amino acid mixtures.
The importance of non-enzymatic browning reactions has not been demonstrated in humification; however, in some special circumstances browning reactions may be important. González-Pérez and others (2004) pointed out that Maillard reactions may take place in soils heated by fires. It should be noted, however, that simple sugars would be expected to be rapidly consumed by microorganisms in natural systems and, therefore, would not be available to participate in browning reactions.
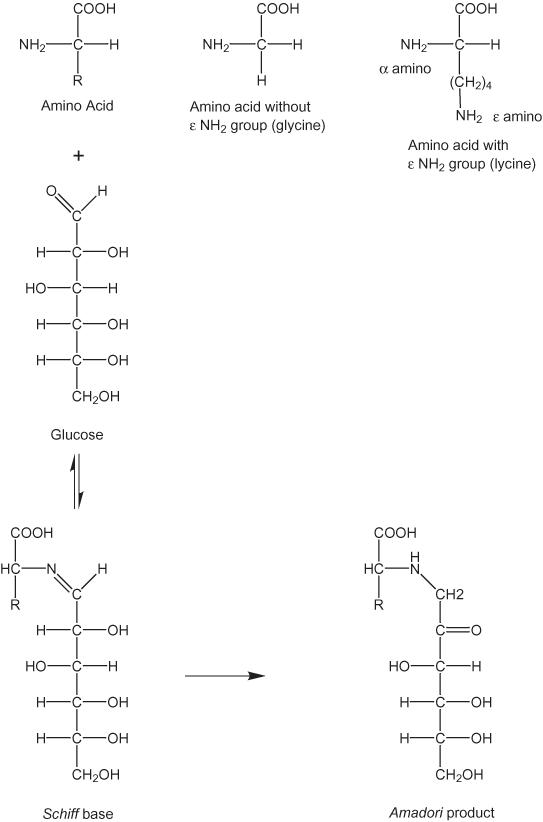
Figure 19. First steps in browning reactions.
Wershaw (1994) proposed that NOM is composed of molecular aggregates of the degradation products of plant tissue. That is to say, humification can be considered as a three-step process of (1) degradation, (2) aggregation of the degradation products, and (3) degradation of the aggregation products. All living organisms are made of molecules that form non-covalently bonded molecular aggregates (supramolecular aggregates). In solution supramolecular aggregation is a reversible process in which the molecules are constantly combining and breaking apart (Lou and others, 2004). In living organisms the various supramolecular aggregates are segregated in a hierarchy of compartments such as organelles, cells, tissues, and so on. However, when an organism dies the barriers between the compartments break down, and their contents are mixed together and degraded. As outlined in the Degradation Pathways of Plant Tissue Components Section, the products of biodegradation of the major components of plant tissue are monomeric or oligomeric units of the polymers that existed in the original organism, and therefore, they can be expected to undergo the same types of non-covalent interactions as the precursor polymers. The polyphenolic compounds (tannins) constitute a notable exception to this rule. They undergo oxidative coupling reactions that produce new types of structures.
During the mixing of the biodegradation products, new supramolecular aggregates form. The process of building these new supramolecular aggregates results in the formation of NOM. In general, with the exception of the polyphenolic compounds, one need not invoke the formation of new covalent bonds in the humification process that leads to the production of NOM.
Wershaw (1994) reviewed observed properties of NOM isolates, and from these data proposed that "* * *the depolymerization and oxidation reactions that take place during the enzymatic degradation of biopolymers produce amphiphiles–molecules that have a polar (hydrophilic) part and a nonpolar (hydrophobic) part. These amphiphiles that result from the partial oxidative degradation of dead biomass assemble spontaneously into ordered aggregates in which the hydrophobic parts of the molecules form the interiors and the hydrophilic parts of the molecules make up the exterior surfaces of the aggregates. These ordered aggregates constitute the humus in soils and sediments. Humus ordered aggregates most likely exist as bilayer membranes coating mineral grains and as micelles in solution." Piccolo (2001) proposed a similar model for humic substances in which they are composed of "* * *supramolecular associations of self-assembling heterogeneous and relatively small molecules derived from the degradation and decomposition of dead biological material." Piccolo (2001) has proposed that supramolecular aggregates are "* * *held together mainly by hydrophobic dispersive forces."
More recent observations of the properties of leaf leachates (Wershaw and others, 1998a,b, and Wershaw and Kennedy, 1998) indicate that hydrolysable and nonhydrolysable tannins are important sources of NOM that were not considered explicitly in the model of Wershaw (1994). However, it now appears that these compounds are of particular importance in understanding the aggregation of NOM because they form strong non-covalent bonds with a variety of different types of compounds and, in particular, with carbohydrates and proteins. Haslam (1998) has reviewed "non-covalent inter- and intra-molecular interactions" of phenolic compounds. Haslam has called these types of interactions "molecular recognition" because of their importance in biochemical processes. Non-covalent interactions in aqueous solutions may be classified into three categories: Lifshitz-van der Waals forces, Lewis acid-base (electron-donor/electron-acceptor) forces, and electrical double-layer forces (van Oss, 2003). The electron donor-acceptor interactions are by far the strongest of these interactions. Included within this category are hydrogen bonds, hydrophobic interactions, ionic interactions, and other polar interactions.
Haslam (1998) pointed out that phenolic compounds are by their very nature amphiphiles because phenolic hydroxyl groups are hydrophilic and aromatic rings are hydrophobic. The amphiphilic character of phenols enhances their interaction with carbohydrates and proteins because the phenolic hydroxyls form hydrogen bonds with the polar groups of carbohydrates and proteins, and the aromatic rings interact with the more hydrophobic parts of the carbohydrate and protein molecules.
Electron-donating or electron-withdrawing substituents on an aromatic ring will polarize the aromatic π electrons. Thus, for example, an alkoxy substituent will cause the ring to exhibit a net negative charge, and conversely, a carbonyl group will cause the ring to exhibit a net positive charge (Cubberley and Iverson, 2001). Oppositely charged rings, therefore, will be attracted both by hydrophobic and electrostatic effects. If a single molecule is composed of aromatic rings that are linked by a flexible polymeric backbone, then hydrophobic interactions will cause the rings to tend to stack one on top of another to form a more compact conformation (foldamer) in solution (Gellman, 1998). If the rings are oppositely charged, then the stacking tendency will be enhanced.
Intermolecular stacked aromatic complexes also form in solution (Haslam, 1998). The association of caffeine with polyphenols has been extensively studied. Cai and others (1990) showed that caffeine molecules are sandwiched between the gallate rings of theaflavin digallate, thereby bridging between theaflavin digallate molecules in solution. Haslam (1998) proposed that the precipitation of some proteins by tannin polyphenols involves "multi-dentate" binding of a protein molecule to a polyphenolic molecule. He pointed out that an average tannin polyphenolic molecule of molecular weight of 1,000 has between 12 and 16 phenolic hydroxyl groups. If both the tannin and protein molecules possess significant conformational mobility then it should be possible to form multiple hydrogen bonds between the phenolic molecules and the proteins molecules. In this scenario, proteins with loose, random conformations in which large numbers of binding sites are exposed would interact more strongly with polyphenols than would compact globular proteins with few exposed binding sites, and this is indeed the case. A similar mechanism would explain the interaction of polyphenols with polysaccharides.
The work of McIntosh and others (1999) provides another example of promotion of aggregation by tannins; they found that hydrolysable tannins (so-called tannic acid) strongly interact with phospholipids in bilayer membranes to bring about aggregation of the bilayer structures. They argue that the tannic acid molecules act as molecular bridges between adjacent bilayers.
The ability of tannin polyphenols to form non-covalent bonds provides an explanation for the preservation of proteins and peptides in soils. NMR studies of soils have demonstrated that almost of all of the nitrogen is in the form of amides (see Knicker, 2002). These findings are supported by the work of Martens and Loeffelmann (2003) who found, using a methanesulfonic acid hydrolysis procedure they developed, that more than 85 percent of the total nitrogen in the soils they analyzed was recovered as amino acids, aminosugars, and ammonia. They concluded that these hydrolysis products are most likely derived from peptides and aminosugars in the soils. Larger polypeptides (proteins) also may be present. The aminosugars are probably in the form of bacterial peptidoglycans. The preservation of those peptides that contain free amino groups is probably due to the formation of protein or peptide-clay complexes (see Wershaw and Pinckney, 1980). Wershaw and Pinckney (1980) showed that deamination of clay-humic complexes released a nitrogen-containing organic phase from the clays. Deamination will eliminate the free amino groups of basic amino acids in proteins and peptides. Laird and others (2001) found that 50 to 66 percent of the total amino acids associated with the fine clay fractions of the agricultural soils they analyzed was arginine. Arginine is the most basic amino acid found in proteins; the guanidinium group of arginine will be positively charged to pH values in excess of 12. Proteins containing arginine or free arginine, therefore, can bond to negatively charged sites of clay surfaces (Laird and others, 2001).
This mechanism cannot be invoked if free amino groups are not present. Zang and others (2000), however, found that humic acid isolates also protect proteins from degradation. They proposed that this protection is brought about by "encapsulation into hydrophobic domains of humic acids." A purely hydrophobic mechanism has been used to explain the sorption of hydrophobic organic compounds in soils and sediments (see Xing, 1998, and references therein); however, proteins are characterized by both polar and nonpolar parts. A more likely encapsulation mechanism would be by hydrogen bonding and hydrophobic interactions of the proteins (or peptides) with the tannin polyphenolic components of NOM. The polyphenols would protect the proteins (peptides) by forming multi-dentate hydrogen-bonded complexes with them (see Haslam, 1998, p. 143-145) and hydrophobic-interaction complexes between the hydrophobic regions of the polyphenols and the proteins. The bound polyphenols would shield the proteins (or peptides) from the action of proteases. Zang and others (2001) found that proteinaceous material from a green alga is preserved from microbial degradation by a highly refractory cell wall component called algaenan.
Carbohydrates also form weak-bonded complexes with polyphenols and proteins. Cai and others (1990) found that hydrogen bonding and hydrophobic interactions are important in the complexation. The sorption of polyphenols by cyclodextrins allows one to study these two types of interactions. The hydrophobic interiors of the cyclodextrins will sorb phenolic groups that are small enough to penetrate the cyclodextrin cavities; phenolic hydroxyl groups interact with the exterior hydroxyl groups of the cyclodextrins (Cai and others, 1990; Sojo and others, 1999). Many polysaccharides such as starch (amylose) exist in coiled conformations that can form host-guest complexes with polyphenols (Cai and others, 1990). Many carbohydrates form hydrogen-bonded complexes with enzymes and other proteins. This binding is often stereospecific, resulting from an intricate network of hydrogen bonds at specific binding sites (Quiocho, 1986).
Etter (1990) has attempted to devise a method for predicting the patterns (morphologies) of hydrogen bonds between proton-donor and proton-acceptor functional groups by studying hydrogen-bond patterns in crystalline solids. She proposed a set of three general rules for hydrogen-bond formation between organic compounds in solution or in the solid state:
With regard to the third rule, many authors have attempted to develop scales that rank compounds with respect to their hydrogen-bond acceptor basicities or hydrogen-bond donor acidities (Haslam, 1998). These scales are not universally applicable because they normally are developed by measuring hydrogen-bond formation constants of a group of proton-donor compounds with an arbitrarily chosen strong proton-acceptor standard or the formation constants of a group of proton-acceptor compounds with an arbitrarily chosen strong proton-donor standard. The relative positions of the donor or acceptor compounds in the scales will be to some extent dependent on the choice of standards and solvents used for the measurements. Anions in general are the strongest proton acceptors and proton-containing cations are the strongest donors (Abraham and others, 1989). Thus, carboxylates, which are the most common anionic functional groups in NOM, can be expected to form especially strong hydrogen-bonded complexes with strong proton donors such as phenols.
As pointed out in the Introduction Section of this report, in the humic polymer model it is postulated that the component molecules of NOM are polymeric species that have been synthesized by secondary synthesis reactions from plant degradation products. It is further postulated that they have unique chemical structures that are different from precursor plant polymers. A large number of these models have been proposed (see Stevenson, 1994, p. 287-294). The models generally have been developed for soil humic acid isolates by taking into account the chemical and physical data that been determined for a particular isolate or group of isolates. The data commonly include functional group distributions, elemental compositions, and number- or weight-average molecular weights.
With the advent of computer modeling (Bruccoleri and others, 2001; Diallo and others, 2001; Schulten and Schnitzer, 1995; Sein and others, 1999), modeling of NOM has reached new levels of mathematical sophistication. The real problem in using these modeling programs is determination of the input data to be used by the program. For example, in the computational scheme used by Diallo and others (2001) the required input data "* * *include: (i) atomic ratios, (ii) functional group ratios, (iii) structural groups centered on carbon atoms and hydrogen atoms, (iv) the nature and amount of molecular fragments and the interfragment bonds, and the number average molecular weight."
In both the manual and computer-aided modeling efforts for soil humic acids, the input data have been obtained by making measurements on humic acid isolates. These isolates are commonly obtained by extraction of soils with 0.1 Molar (M) NaOH and precipitation of the humic acid by adjusting the solution pH to between 1 and 2. Different purification steps to remove minerals matter also may be used. The resulting isolates are complex mixtures of different molecular species (Wershaw and Pinckney, 1973; Wershaw, 1994). This is not at all surprising in light of the fact that soil organic matter is composed of partially degraded plant tissue, biomass from microorganisms, coatings on mineral grains, pyrolytic carbon, organic precipitates, and DOM in soil water (Cambardella and Elliott, 1992 and 1994; Six and others, 2001; Sohi and others, 2001; Wershaw, 2000). Simpson and others (2002) demonstrated by multi-dimensional NMR analysis of reverse-phase chromatographic fractions that NaOH extracts of soils are composed of mixtures of "* * *aliphatic acids, ethers, esters and alcohols; aromatic lignin derived fragments; polysaccharides and polypeptides."
Sein and others (1999) have attempted to model a "humic acid building block" and the stereochemistry of this "building block." The evidence cited in the previous paragraph that humic acids are complex mixtures precludes the existence of such a "building block." That is to say, humic acid is not a single, identifiable entity. Similar objections can be made to all of the other polymer models that have been proposed.
The molecular aggregate models that have been proposed by Wershaw (1994) and Piccolo (2001) are better representations of the actual state of NOM in soil extracts than the humic polymer models; however, they provide only a very generalized idea of the structure of NOM aggregates in soils prior to extraction. As pointed out in the previous section, soil NOM consists of partially degraded plant tissue, biomass from microorganisms, organic coatings on mineral grains, pyrolytic carbon, organic precipitates, and DOM in soil water; for want of a better term, each of these will be called a separate NOM compartment. A similar set of compartments exists in sediments. The NOM in each one of these compartments has a different composition. Unfortunately, the usual practice of extracting NOM from a whole soil yields a solution in which the components of NOM from all of these compartments that are soluble in the extracting solvent system are mixed together.
The concept of NOM compartments described above is somewhat analogous to the phase concept in thermodynamic. A phase is a homogeneous region in a thermodynamic system. Only the DOM compartment is truly a phase. The other compartments are not homogeneous regions; however, they are identifiable regions that can be separated from soils or sediments. Each of these compartments is composed of a set of components (chemical substances); some of these components are inorganic and some organic. The organic components constitute the NOM in the compartment. These components will, in general, interact with each other and the inorganic components in the compartment. As in thermodynamics, one must identify the components and their interactions in each phase or compartment before attempting to study the entire system, whether it be a soil or a well-defined system of two or three components.
Separation and characterization of soil or sediment NOM (SOM) from each of the compartments (partially degraded plant tissue, biomass from microorganisms, organic coatings on mineral grains, pyrolytic carbon, organic precipitates, and DOM in soil water) should allow one to develop more representative models for NOM supramolecular aggregates in soil and sediment systems. This is especially important because the NOM in each of these compartments can be expected to have different sorption, ion exchange, and buffering properties.
A start has been made on techniques to achieve these separations. A number of groups have fractionated soils by particle size and density, and have characterized the NOM in these fractions with solid-state 13C NMR (Baldock and others, 1992; Chen and Chiu, 2003; Golchin and others, 1997; Kahle and others, 2003; Laird and others, 2001; Preston and others, 2000; Schmidt and Kögel-Knabner, 2002; Six and others, 2001; Sohi and others, 2001). Solid-state 13C NMR provides a nondestructive means of characterizing the NOM in soil fractions without alteration.
The physical separation techniques used by the various groups were designed to isolate the size and density fractions with minimal alteration of the NOM. The procedure used by Golchin and others (1997) is a typical density fractionation procedure. In this procedure 20-gram (g) samples from Japanese volcanic ash soils (andosols) were suspended in 200 milliliters (mL) of sodium polytungstate solution of density 1.6 grams/cubic centimeter (g/cc) in capped 250 mL glass centrifuge tubes by inverting the tube gently five times by hand. The tubes were allowed to stand for 30 minutes (min) and was then centrifuged for 30 min at 2,000 revolutions per minute (rpm). The floating particles and the supernatant solution were decanted, and the particles were isolated by filtration and washed to remove residual polytungstate. The particles were dried and weighed; this fraction was designated the free light fraction with a density (d) less than (<) 1.6 g/cc. The residual soil from the first floatation was resuspended in 80 mL of 1.6 g/cc solution and sonicated in an ice bath for 5 min. The volume was then adjusted to 200 mL with polytungstate solution and centrifuged, filtered, and washed as before. The fraction isolated was called the occluded d < 1.6 g/cc fraction. The residual soil was further fractionated by sequential suspension in 1.8 g/cc and 2.0 g/cc solutions. In this way, fractions with densities between 1.6–1.8 g/cc and 1.8–2.0 g/cc and greater than (>) 2 g/cc were isolated. A free fraction isolated by floatation with pure water also was isolated; it was designated as the free d < 1.0 g/cc fraction.
Size fractionation by dry sieving also has been used on soil samples; the dry sieving may be followed by wet sieving to isolate smaller particle-size fractions. Clay fractions are generally isolated by dispersing sieved soils in water and allowing the different clay size particles to settle from suspension. Kahle and others (2003) isolated coarse and fine clay fractions from illitic soils (soils in which illite was the major clay mineral in the soils). NOM was in the form of coatings on the clay surfaces.
The different soil fractions of Golchin and others (1997) represent different stages in the diagenesis of NOM. The least degraded material was isolated in the free d < 1.0 g/cc and the free d < 1.6 g/cc fractions; they yielded NMR spectra very similar to those of fresh plant litter on soil surfaces. Carbohydrate bands are the dominant ones in these spectra. In the occluded d < 1.6 fractions and the more dense fractions, the relative intensities of the carbohydrate bands were reduced in comparison to bands attributable to alkyl hydrocarbons and aromatic moieties. A similar reduction in the relative intensities of carbohydrate bands was observed in the solid-state 13C NMR of litter samples that had undergone weathering for different lengths of time on the floor of a pine forest (Qualls and others, 2003); Qualls and others (2003) observed that the longer the weathering period, the greater the loss of carbohydrates from the litter. Preston and others (1998) also observed a loss of carbohydrates brought about by decay in coarse woody debris in coastal forests of British Columbia. Nierop and others (2001) found lower concentrations of carbohydrates in the NOM in soils than in the overlying litter or fresh plant material from a primary vegetation succession (algae, moss, heather, and pine) in the central Netherlands. Accumulation of resistant aliphatic polymers derived from mosses also was detected in the soils. They found that lignin degraded more rapidly than tannins.
Soil microbial biomass is difficult to isolate and characterize; however, Golchin and others (1996) and Baldock and others (1990) have used an indirect method to characterize soil biomass. Baldock and others (1990) isolated bacteria and fungi from a sandy loam soil. The isolates were amended with glucose and the solid-state 13C NMR spectra of the cultures were measured. They found that the spectra from the two materials were distinctive. The bacterial material contained more alkyl, aromatic, and carboxyl groups and less O-alkyl and acetal groups than the fungal material. They compared these NMR spectra with spectra of soil fractions incubated with 13C-enriched glucose and concluded that "* * *the soil microbial population was dominated by fungi." Golchin and others (1996) extended this work by inoculating artificial soils with soil microorganisms and incubating the artificial soils with unlabelled and 13C-labelled glucose. They found that 24 to 25 percent of the carbon was in the form of alkyl groups, 56 to 63 percent in the form of O-alkyl groups, 11 to 15 percent in the form of carbonyl groups, and a small amount in aromatic and olefinic groups. By using proton-spin relaxation editing techniques they were able to distinguish between slowly relaxing (long T1(H)) functional groups and rapidly relaxing (short T1(H)) functional groups. Alkyl carbons in long- and short-chain structures had long T1(H) values as did N-alkyl groups. Carbonyl carbon atoms and O-alkyl groups had short T1(H) values. Golchin and others (1996) concluded that the alkyl carbon groups were attached to clay mineral surfaces by polysaccharide mucilages and that the O-alkyl groups represented neutral and acidic extracellular saccharides. Lichtfouse and others (1998) also found evidence for straight-chain aliphatic biopolymers that are resistant to degradation in soils. This resistance to degradation may be due to attachment of the aliphatic biopolymers to clay minerals.
Kahle and others (2003) measured the solid-state 13C NMR spectra of fine and coarse clay fractions from soils from seven different sites. They found that the fine clay fractions had more amide carbonyl carbon and less carbohydrate carbon than the corresponding coarse clay fractions. Kahle and others (2003) pointed out that NOM may also be associated with iron oxides in the fine clay fractions.
The presence of iron oxides or other paramagnetic materials in a sample presents special problems for NMR analysis (Smernik and Oades, 2002). If the concentration of the paramagnetic species is similar to that of the carbon content of the sample, then severe broadening of the 13C NMR bands will occur. Gonçalves and others (2003) have developed a procedure using hydrofluoric acid to remove iron oxides from soil samples prior to NMR analysis. Carbon functional group distributions do not appear to be altered significantly by their procedure.
Wershaw and others (1996) showed that compost leachate DOM formed bilayer membrane-like coatings on alumina surfaces. The experiments conducted by Wershaw and others (1996) were designed to simulate what happens when leachate from decomposing plant litter is sorbed by hydrous metal oxides in soils. Alumina was chosen as a representative hydrous metal oxide because it is not paramagnetic, and therefore, NMR can be used to characterize the sorbed organic material. In addition, alumina surfaces are present in clay minerals. Hydrous metal oxide surfaces are composed of hydroxyl ions bonded to metal ions. Wershaw and others (1996) showed that organic anionic species from the compost leachate replaced hydroxyl ions to form inner sphere complexes with aluminum ions. Some of the anions from the compost leachate adsorbed to the alumina surface were amphiphiles. Wershaw and others (1996) proposed that the amphiphile molecules (such as long-chain aliphatic acids) "* * *are adsorbed on metal oxide surfaces through a combination of polar and hydrophobic interaction mechanisms. This combination of polar and hydrophobic mechanisms will lead to the formation of bilayer coatings of the leachate amphiphilic molecules on the oxide surfaces."
Sorption of NOM also takes place on clay mineral surfaces. These surfaces oftentimes are negatively charged because of isomorphous cation substitution (Sposito and others, 1999). As pointed out above, basic amino acids and peptides, proteins, and amino sugars containing basic amino acids are bound to negatively charged clay mineral surfaces. Wershaw and Pinckney (1980) characterized a colloidal clay-humic complex that had been isolated as a fraction of a soil humic acid. Wershaw and others (1988 and 1990) used liquid-state 13C NMR to characterize colloidal clay-humic complexes isolated from different soils and sediments. They found that NMR spectra of many of these clay-complexes consisted of very sharp carbohydrate bands. These bands are most likely due to the presence of amino sugars or peptidoglycans that are bound to the clay surfaces by basic amino acid moieties.
Techniques for the isolation and characterization of pyrolytic carbon (so-called black carbon) in soils and sediments have not been well developed. Golchin and others (1997) found by light microscopy that some of the density fractions they isolated were enriched in black carbon; however, other soil components also were present in the fractions. Skjemstad and others (2002) used a series of physical and chemical treatments to isolate charcoal from agricultural soils. Unfortunately, a high-energy photo-oxidation step was included in their procedure to remove "more labile fractions of organic matter"; this step could very well alter existing charcoal or produce black carbon from the so-called labile organic matter. Another highly aggressive chemical oxidation procedure using dichromate and sulfuric acid has been used to isolate black carbon from sediments (Masiello and Druffel, 1998). This procedure can be expected to markedly alter the black carbon. Recently, a group of scientists convened a meeting at the Friday Harbor Laboratories of the University of Washington called "New Approaches in Marine Organic Biogeochemistry". Part of the meeting was devoted to developing black-carbon reference materials (Schmidt and others, 2003). Hopefully, efforts of this nature should lead to better isolation and characterization techniques.
Rutherford (D.W. Rutherford, U.S. Geological Survey, written commun., 2004) has prepared wood charcoals under conditions similar to those that prevail during forest fires. He measured the surface areas, elemental compositions, and NMR and IR spectra of charcoals prepared by heating pine and poplar wood at different temperatures for different time durations. Future studies are planned to compare the charcoals prepared under controlled conditions with charcoals collected from areas that have undergone forest fires.
Precipitation of NOM in soils can take place during evaporation of soil solutions. Metal ion bridging between NOM molecules also will bring about precipitation of NOM. At the present time (2004), no techniques exist for the isolation of organic precipitates from soils. Natural precipitates, however, can be simulated by adding selected metal ions to interstitial solutions collected from soils or by evaporating these solutions.
Merdy and others (2000) and Guillon and others (2001) attempted to simulate the interaction of metal ions with NOM by studying the binding of Cu (II), Mn(II), and Fe(III) with lignin monomeric, dimeric, and polymeric model compounds. These metals formed strong complexes with the lignin model compounds. Guillon and others (2001) proposed that part of the Fe(III) in soils will be immobilized by complexation with insoluble lignin components, and that part of Fe(III) will be mobilized by complexation with soluble lignin components. They further proposed that sorbed Fe (III) will promote oxidation of the NOM. The studies of Merdy and others (2000) and Guillon and others (2001) could easily be extended to the interaction of metal ions with DOM components isolated from soil interstitial solutions in order to provide a more realistic simulation of the actual interactions in soil-water systems.
Soil solutions may be readily collected using different types of lysimeters (see Charbeneau and Daniel, 1992, and the references therein). The pressure-vacuum lysimeter is the most commonly used type. It consists of a porous cup that is placed in a borehole; the space between the cup and the inside of the borehole is filled with a fine silica powder. The cup is evacuated with a vacuum pump to draw water from the soil into the cup. Qualls and Haines (1992) and Hagedorn and others (2004) used these devices to collect soil solutions. Qualls and Haines (1992) found that in oak and hickory forests of the southern Appalachian Mountains large quantities of DOM are generated on the forest floors. They have interpreted their data as indicating that most of the DOM is rapidly adsorbed in the A horizons of the forest soils; a small amount of DOM remains in solution. It appears that biodegradation in the solution phase is too slow to remove much DOM from the soil solutions; however, the increased concentration of NOM on the soil particles appears to favor biodegradation. Hagedorn and others (2004) have interpreted the data they collected from 13C tracer studies in model beech and spruce forest ecosystems in a somewhat different way. Their results suggest that organic carbon compounds freshly leached from forest floor litter are rapidly degraded to carbon dioxide, and that only a small amount remains in solution. The DOM in the soil solutions collected at 5-10 cm depth "* * *originated mainly from native soil C and pools of humified C smaller than 50 µm." They further point out "* * *that dissolved organic C is produced during incomplete decomposition of recalcitrant soil C, while easily degradable components are rapidly consumed by soil microbes and thus make minor contributions to the dissolved organic C." Neither Qualls and Haines (1992) nor Hagedorn and others (2004) have characterized the DOM in their soil solutions. Nierop and Buurman (1999) used solid-state 13C NMR to characterize the total DOM in water extracts of podzol soils; however, they did not fractionate their extracts. Fractionation would provide a more detailed characterization of the soil-water composition.
Leenheer and others (2000) have developed the most comprehensive fractionation and characterization procedure for DOM that has yet been published. This procedure yields five different fractions: (1) a colloidal fraction, (2) a hydrophobic NOM fraction, (3) a fraction of intermediate polarity (called transphilic NOM), (4) a hydrophilic acids plus neutrals NOM fraction, and (5) a hydrophilic base NOM fraction. Leenheer and others (2000) used this procedure to characterize the DOM in several drinking-water supplies. They found that the colloidal fractions isolated from these waters consisted mainly of N-acetyl amino sugars; the hydrophobic fractions were similar to the fulvic acids that are commonly isolated from natural waters by resin adsorption. They further found that "As the hydrophilic character of the fraction increases, carbon percentage decreases and nitrogen percentage increases." Barber and others (2001) have applied this procedure with some modifications to characterization of DOM in water-treatment wetlands. This procedure can be used for the characterization of soil-water DOM.
The work of Leenheer and others (2000) indicates that DOM operationally defined by passage through a 0.4 micrometer filter actually consists of two compartments: (1) truly dissolved DOM and (2) colloidal DOM. The colloid DOM compartment would include not only the high molecular weight organic colloids that Leenheer and others (2000) isolated, but also NOM adsorbed to colloidal mineral particles.
Evidence exists that DOM in natural waters also forms supramolecular aggregates. Using polarography Hunter and Lee (1986) found that humic acid isolated from a natural water was four times more surface active than any of the other organic components in the water. When the humic acid and the other organic components were mixed at natural concentrations the surface activity of the mixture was only about 50 percent of the sum of the surface activities of the individual components. They interpreted the results of the work as indicating that the DOM components form aggregates in solution. Wershaw and Hayes (2001) have reviewed the literature on solubilization of anthropogenic compounds by DOM. They concluded that the enhanced solubilities of hydrophobic organic compounds that have been observed in natural waters are due to partitioning of the molecules of the hydrophobic compounds into premicellar DOM aggregates. Premicellar aggregates are those that form at concentrations lower than the critical micelle concentration (CMC) for the amphiphilic molecules under consideration. For example, Zimmels and Metzer (1976) found evidence for the formation of premicellar oleate aggregates at concentrations as low as 0.01 CMC. Aggregation of amphiphilic molecules (molecules that have separate hydrophilic and hydrophobic parts) is caused by hydrophobic interactions between the hydrophobic parts of the molecules. Polyvalent cations also can bring about intermolecular aggregation by bridging between polar groups of two or more molecules (Gamble and others, 1984). Another possible aggregation mechanism for DOM is by interaction of tannins with protein and carbohydrate components of DOM as discussed in the Interactions of Degradation Products Section. Pacheco and others (2003) studied the interactions of solutions of soil, peat, Leonardite, and coal humic acids with a variety of different organic and inorganic xenobiotic compounds. They concluded that some of these compounds form supramolecular complexes with the humic acid molecules. They proposed that ion binding, hydrogen bonding, van der Waals forces, ligand exchange, hydrophobic and hydrophilic adsorption, charge-transfer complexation, and sequestration accounted for the aggregation they observed.
Agricultural scientists have recognized the importance of natural organic matter (NOM) in enhancing soil fertility, soil structure, and water-holding capacity for more than 200 years. More recently, its importance as a carbon sink in the global carbon cycle has been recognized. NOM is generally divided into two different pools: (1) soil organic matter (SOM) and dissolved organic matter (DOM) with the SOM pool being the larger of the two. Early workers understood that SOM is formed mainly from the degradation products of dead plant tissue with a much lesser contribution from decaying animal remains. Most of these early workers assumed that SOM was composed of the end products of synthetic reactions that alter the structures of plant degradation products. Other workers, however, maintained that SOM is a complex mixture of plant degradation products. This controversy has persisted to the present day, and a similar controversy exists with regard to DOM.
This controversy has led to the development of two different types of conceptual models for NOM: (1) the humic polymer model and (2) the molecular aggregate model. The belief that the component molecules of NOM are produced by secondary synthesis reactions from degradation products has led to models in which the NOM molecules are depicted as large (humic) polymers with unique chemical structures that are different from those of starting materials. The proposal that NOM is composed of the partial degradation products of plant polymers has resulted in the development of models in which NOM is composed of molecular aggregates (supramolecular aggregates) of the degradation products held together by non-covalent bonds.
The preponderance of evidence favors the molecular-aggregate models. The molecular-aggregate models were developed by studying the properties of NOM extracted from soils and natural waters, and, as such, they provide only a very generalized picture of the structure of NOM aggregates in soils, sediments, and natural waters prior to extraction. A compartmental model would be more representative of NOM in soils and sediments. In this model NOM is divided into the following compartments: (1) partially degraded plant tissue, (2) biomass from microorganisms, (3) organic coatings on mineral grains, (4) pyrolytic carbon, (5) organic precipitates, and (6) DOM in water. Within each of these compartments there are NOM supramolecular aggregates that will be dissolved by the solvent systems that are used for extraction of NOM from soils and sediments. In order to better understand the interactions of NOM in soils and sediments, the NOM in each of these components must be studied separately. This is especially important because the NOM in each of these compartments can be expected to have different sorption, ion exchange, and buffering properties.
DOM as normally isolated actually consists of two compartments: (1) truly dissolved DOM and (2) colloidal DOM. The colloid DOM compartment consists of high molecular weight organic colloids and NOM adsorbed to colloidal mineral particles. There is evidence that DOM molecules also form supramolecular aggregates in natural waters.
Abraham, M.C., Duce, P.P., Prior, D.V., Barratt, D.G., Morris, J.J., and Taylor, P.J., 1989, Hydrogen bonding. Part 9. Solute proton donor and acceptor scales for use in drug design: Journal of the Chemical Society, Perkin Transactions 2, p. 1355–1375.
Albersheim, P., Darvill, A.G., O'Neill, M.A., Schols, H.A., and Voragen, A.G.J., 1996, An Hypothesis: The same six polysaccharides are components of the primary cell walls of all higher plants, in Visser, J. and Voragens, A.G.J., eds., Progress in biotechnology 14—Pectins and pectinases: Amsterdam, Elsevier, p. 47–55.
Anderson, Traute-Heidi, and Domsch, K.H., 1993, The metabolic quotient for CO2 (qCO2) as a specific activity parameter to assess the effects of environmental conditions, such as pH, on the microbial biomass of forest soils: Soil Biology and Biochemistry, v. 25, p. 393–395.
Angers, D.A., N'dayegamiye, A., and Côté, D., 1993, Tillage-induced differences in organic matter of particle-size fractions and microbial biomass: Soil Science Society of America Journal, v. 57, p. 512–516.
Atalla, R.H., 1999, Celluloses, in Pinto, B.M., ed., Comprehensive natural products chemistry, vol. 3, Carbohydrates and their derivatives including tannins, cellulose, and related lignins: Amsterdam, Elsevier, p. 530–598.
Atalla, R.H. and VanderHart, D.L., 1999, The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses: Solid State Nuclear Magnetic Resonance, v. 15, p.1–19.
Baldock, J.A., Oades, J.M., Waters, A.G., Ping, X., Vassallo, A.M., and Wilson, M.A., 1992, Aspects of the chemical structure of soil organic materials as revealed by solid-state 13C NMR spectroscopy: Biogeochemistry, v. 16, p. 1–42.
Baldock, J.A., Oades, J.M., Vassallo, A.M., and Wilson, M.A., 1990, Solid-state CP/MAS 13C N.M.R. analysis of bacterial and fungal cultures isolated from a soil incubated with glucose: Australian Journal of Soil Research, v. 28, p. 213–225.
Barber, L.B., Leenheer, J.A., Noyes, T.I., and Stiles, E.A., 2001, Nature and transformation of dissolved organic matter in treatment wetlands: Environmental Science and Technology, v. 35, p. 4805–4816.
Baruah, A.M., and Mahanta, P.K., 2003, Fermentation characteristics of some Assamica clones and process optimization of black tea manufacturing: Journal of Agricultural and Food Chemistry, v. 51, p. 6578–6588.
Beldman, G., Mutter, M., Searle-van Leeuwen, M.J.F., van den Broek, L.A.M., Schols, H.A., aned Voragen, A.G.J., 1996, New enzymes active toward pectic structures, in Visser, J. and Voragens, A.G.J., eds., Progress in biotechnology 14—Pectins and pectinases: Amsterdam, Elsevier, p. 231–245.
Benen, J.A.E., Kester, H.C.M., Parenicová, Lucie; and Visser, Jaap, 1996, Kinetics and mode of action of Aspergillus niger polygalacturonases in Visser, J. and Voragens, A.G.J., eds., Progress in biotechnology 14—Pectins and pectinases: Amsterdam, Elsevier, p. 221–230.
Bergmann, C.W., Cook, B., Darvill, A.G., Albersheim, P., Bellincampi, D., and Caprari, C., 1996, The effect of glycosylation of endopolygalacturonases and polyglacturonase inhibiting proteins on the production of oligogalacturonides, in Visser, J. and Voragens, A.G.J., eds., Progress in biotechnology 14—Pectins and pectinases: Amsterdam, Elsevier, p. 275–282.
Bernards, M.A., 2002, Demystifying suberin: Canadian Journal of Botany, v. 80, p. 227–240.
Bernards, M.A. and Lewis, N.G., 1998, The macromolecular aromatic domain in suberized tissue—A changing paradigm: Phytochemistry, v. 47, p. 915–933.
Berto, Philippe; Comménil, Pascal; Belingheri, Lionel; Dehorter, Bertrand, 1999, Occurrence of a lipase in spores of Alternaria brassicicola with a crucial role in the infection of cauliflower leaves: FEMS (Federation of European Microbiological Societies) Microbiology Letters, v. 180, p. 183–189.
Brinkmann, Thomas; Hörsch, Philip; Sartorius, Daniel, and Frimmel, F.H., 2003, Photoformation of low-molecular-weight organic acids from brown water dissolved organic matter: Environmental Science and Technology, v. 37, p. 4190–4198.
Brown, R.G., 1999, Emission and flash techniques in environmental photochemistry in Boule, P., ed., The handbook of environmental chemistry v. 2 part L, Environmental photochemistry: Berlin, Springer-Verlag, p. 27–61.
Bruccoleri, A.G., Sorenson, B.T., and Langford, C.H., 2001, Molecular modeling of humic substances, in Ghabbour, E.A. and Davies, Geoffrey, eds., Humic substances—Structures, models and functions: Cambridge, Royal Society of Chemistry, p. 193–208.
Burlat, Vincent; Kwon, Mi; Davin, L.B., and Lewis, N.G., 2001, Dirigent proteins and dirigent sites in lignifying tissues: Phytochemistry, v. 57, p. 883–897.
Cai, Ya; Gafney, S.H., Lilley, T.H., Magnolato, Daniele; Martin, Russell; Spencer, C.M., and Haslam, Edwin, 1990, Polyphenol interactions. Part 4. Model studies with caffeine and cyclodextrins: Journal of the Chemical Society, Perkin Transactions 2, 1990, p. 2197–2208.
Camarero, Susana; Galletti, G.C., and Martínez, A.T., 1994, Preferential degradation of phenolic lignin units by two white rot fungi: Applied and Environmental Microbiology, v. 60, p. 4509–4516.
Cambardella, C.A., and Elliott, E.T., 1992, Particulate soil organic-matter changes across a grassland cultivation sequence: Soil Science Society of America Journal, v. 56, p. 777–783.
Cambardella, C.A., and Elliott, E.T., 1994, Carbon and nitrogen dynamics of soil organic matter fractions from cultivated grassland soils: Soil Science Society of America Journal, v. 58, p. 123–130.
Charbeneau, R.J. and Daniel, D.E., 1992, Contaminant transport in unsaturated flow, in Maidment, D.R., Handbook of hydrology: New York, McGraw-Hill, Inc., p. 15.1–15.46.
Chen, Jenn-Shing and Chiu, Chih-Yu, 2003, Characterization of soil organic matter in different particle-size fractions in humid subalpine soils by CP/MAS 13C NMR: Geoderma, v. 117, p. 129–141.
Claeys, Magda; Graham, Bim; Vas, Gyorgy; Wang, Wu; Vermeylen, Reinhilde; Pashynska, Vlada; Cafmeyer; Guyon, Pascal; Andreae, M.O., Artaxo, Paulo; and Maenhaut, Willy, 2004, Formation of secondary organic aerosols through photooxidation of isoprene: Science, v. 303, p. 1173–1176.
Crestini, Claudia; Bernini, Roberta; Porri, Antonio; and Giovannozzi-Sermanni, Geivanni, 1996, Biodegradation of monomeric, dimeric and polymeric lignin models by Lentinus edodes: Holzforschung, v. 50, p. 193–200.
Crestini, Claudia; Sermanni, G.G., and Argyropoulos, D.S., 1998, Structural modifications induced during biodegradation of wheat lignin by Lentinula edodes: Bioorganic & Medicinal Chemistry, v. 6, p. 967–973.
Cubberley, M.S., and Iverson, B.L., 2001, 1H NMR investigation of solvent effects in aromatic stacking interactions: Journal of the American Chemical Society, v. 123, p. 7560–7563.
Dangl, J.L., Dietrich, R.A., and Thomas, Howard, 2000, Senescence and programmed cell death, in Buchanan, B.B., Gruissen, Wilhelm; and Jones, R.L., eds., Biochemistry and molecular biology of plants: Rockville, Maryland, American Society of Plant Physiologists, p. 1044–1100.
Davies, A.P., Goodsall, Chris; Cai, Ya; Davis, A.L., Lewis, J.R., Wilkins, John; Wan, Xiaochun; Clifford, M.N., Powell, Chris; Parry, Andrew; Thiru, Ambalavanar; Safford, Robert; and Nursten, H.E., 1999, Black tea dimeric and oligomeric pigments—Structures and formation, in Gross, G.G., Hemingway, R.W., Yoshida, Takashi; and Branham, S.J., eds., Plant polyphenols 2—Chemistry, biology, pharmacology, ecology: New York, Kluwer Academic/Plenum Publishers, p. 697–723.
Dec, Jerzy; Haider, Konrad; and Bollag, Jean-Marc, 2003, Release of substituents from phenolic compounds during oxidative coupling reactions: Chemosphere, v. 52, p. 549–556.
Diallo, M.S., Faulon, Jean-Loup; Goddard, W.A., III, and Johnson, J.H., Jr., 2001, Binding of hydrophobic organic compounds to dissolved humic substances, A predictive approach based on computer assisted structural elucidation, atomistic simulations and Flory-Huggins solution theory, in Ghabbour, E.A. and Davies, Geoffrey, eds., Humic substances—Structures, models and functions: Cambridge, Royal Society of Chemistry, p. 221–237.
Donaldson, L.A., 2001, Lignification and lignin topochemistry—an ultrastructural view: Phytochemistry, v. 57, p. 859–873.
Douglas, C.J., 1996, Phenylpropanoid metabolism and lignin biosynthesis, From weeds to trees: Trends in Plant Science, v. 1, p. 171–178.
Eschenfeldt, W.H., Zhang, Yeyan; Samaha, Hend; Stols, Lucy; Eirich, L.D., Wilson, C.R., and Donnelly, M.I., 2003, Transformation of fatty acids catalyzed by cytochrome P450 monooxygenase enzymes of Candida tropicalis: Applied and Environmental Microbiology, v. 69, p. 5992–5999.
Etter, M.C., 1990, Encoding and decoding hydrogen-bond patterns of organic compounds: Accounts of Chemical Research, v. 23, p. 120–126.
Fairley, D.J., Boyd, D.R., Sharma, N.D., Allen, C.C.R., Morgan, P., and Larkin, M.J., 2002, Aerobic metabolism of 4-hydroxybenzoic acid in Archaea via an unusual pathway involving an intramolecular migration (NIH Shift): Applied and Environmental Microbiology, v. 68, p. 6246–6255.
Faust, B.C., 1999, Aquatic photochemical reactions in atmospheric, surface, and marine waters—Influences on oxidant formation and pollutant degradation, in Boule, P., ed., The handbook of environmental chemistry v. 2 part L, Environmental photochemistry: Berlin, Springer-Verlag, p. 101–122.
Ferreira, Daneel; Nel, R.J.J., and Bekker, Riaan, 1999, Condensed Tannins, in Pinto, B.M., ed., Comprehensive natural products chemistry, v. 3, Carbohydrates and their derivatives including tannins, cellulose, and related lignins: Amsterdam, Elsevier, p. 747–797.
Friedman, Mendel, 1996, Food browning and its prevention, An overview: Journal of Agricultural and Food Chemistry, v. 44, p. 631–653.
Gamble, D.S., Langford, G.H., and Underdown, A.W., 1984, The interrelationship of aggregation and cation binding of fulvic acid, in Kramer, C. and Duinker, J.C., eds., Complexation of trace metals in natural waters: The Hague, Martinus Nijhoff/Dr. W. Junk, p. 349–356.
Gélinas, Yves; Prentice, K.M., Baldock, J.A. and Hedges, J.J., 2001, An improved thermal oxidation method for the quantification of soot/graphitic black carbon in sediments and soils: Environmental Science and Technology, v. 35, p. 3519–3525.
Gellman, S.H., 1998, Foldamers, A manifesto: Accounts of Chemical Research, v. 31, p. 173–180.
Glaser, Bruno; Haumaier, Ludwig; Guggenberger, Georg; and Zech, Wolfgang, 2001, The 'Terra Preta' phenomenon, A model for sustainable agriculture in the humid tropics: Naturwissenschaften, v. 88, p. 37–41.
Golchin, A., Baldock, J.A., Clarke, P., Higashi, T., and Oades, J.M., 1997, The effects of vegetation and burning on the chemical composition of soil organic matter in volcanic ash soil by 13C NMR spectroscopy. II. Density fractions: Geoderma, v. 76, p. 175–192.
Golchin, A., Clarke, P., and Oades, J.M., 1996, The heterogeneous nature of microbial products as shown by solid-state 13C CP/MAS NMR spectroscopy: Biogeochemistry, v. 34, p. 71–96.
Goldstone, J.V., Pullin, M.J., Bertilsson, S., and Voelker, B.M., 2002, Reactions of hydroxyl radical with humic substances—Bleaching, mineralization, and production of bioavailable carbon substrates: Environmental Science and Technology, v. 36, p. 364–372.
Gonçalves, C.N., Dalmolin, R.S.D., Dick, D.P., Knicker, Heike; Klamt, Egon; Kögel-Knabner, Ingrid, 2003, The effect of 10% HF treatment on the resolution of CPMAS 13C NMR spectra and on the quality of organic matter in ferralsols: Geoderma, v. 116, p. 373–392.
González-Pérez, J.A., González-Vila, F.J., Almendros, Gonzalo; and Knicker, Heike, 2004, The effect of fire on soil organic matter—A review: Environment International, v. 30, p. 855–870.
Gregory, Abigail, and Bolwell, G.P., 1999, Hemicelluloses, in Pinto, B.M., ed., Comprehensive natural products chemistry, v. 3, Carbohydrates and their derivatives including tannins, cellulose, and related lignins: Amsterdam, Elsevier, p. 599–615.
Gross, G.G., 1999, Biosynthesis of hydrolysable tannins, in Pinto, B.M., ed., Comprehensive natural products chemistry, v. 3, Carbohydrates and their derivatives including tannins, cellulose, and related lignins: Amsterdam, Elsevier, p. 799–826.
Guenther, Alex; Hewitt, C.N., Erickson, David; Fall, Ray; Geron, Chris; Graedel, Tom; Harley, Peter; Klinger, Lee; Lerdau, Manuel; McKay, W.A., Pierce, Tom; Scholes, Bob; Steinbrecher, Rainer; Tallamraju, Raja; Taylor, John; Zimmerman, Pat, 1995, A global model of natural volatile organic compound emissions: Journal of Geophysical Research, v. 100 (D5), p. 8873–8892.
Guillon, Emmanuel; Merdy, Patricia; Aplincourt, Michel; Dumonceau, Jacques; and Vezin, Hervé, 2001, Structural characterization and iron (III) binding ability of dimeric and polymeric lignin models: Journal of Colloid and Interface Science, v. 239, p. 39–48.
Hagedorn, F., Saurer, M., and Blaser, P., 2004, A 13C tracer study to identify the origin of dissolved organic carbon in forested mineral soils: European Journal of Soil Science, v. 55, p. 91–100.
Harder, Jens and Probian, Christina, 1995, Microbial degradation of monoterpenes in the absence of molecular oxygen: Applied and Environmental Microbiology, v. 61, p. 3804–3808.
Harwood, C.S. and Parales, R.E., 1996, The β-ketoadipate pathway and the biology of self-identification: Annual Reviews of Microbiology, v. 50 p. 553–590.
Haslam, Edwin, 1995, Secondary metabolism—Evolution and function, Products or processes?: Chemoecology, v. 5/6, p. 89–95.
Haslam, Edwin, 1998, Practical polyphenolics—From structure to molecular recognition and physiological action: Cambridge, U.K., Cambridge University Press, 422 p.
Haslam, Edwin, 2003, Thoughts on thearubigins: Phytochemistry, v. 64, p. 61–73.
Hatcher, P.G., and Spiker, E.C., 1988, Selective degradation of plant biomolecules, in Frimmel, F.H. and Christman, R.F., eds., Humic substances and their role in the environment: Chichester, U.K., John Wiley & Sons, p. 59–74.
Heredia, Antonio, 2003, Biophysical and biochemical characteristics of cutin, A plant barrier biopolymer: Biochemica et Biophysica Acta, v. 1620, p. 1–7.
Hintner, Jan-Peter; Lechner, Christa; Riegert, Ulrich; Kuhm, A. E., Storm, Thomas; Reemtsma, Thorsen; and Stolz, Andreas, 2001, Direction ring fission of salicylate by a salicylate 1,2 dioxygenase activity from Pseudaminobacter salicylatoxidans: Journal of Bacteriology, v. 183, p. 6936–6942.
Hoondal, G.S., Tiwari, R.P., Tewari, R., Dahiya, N., and Beg, Q.K., 2002, Microbial alkaline pectinases and their industrial applications: a review: Applied Microbiology and Biotechnology, v. 59, p. 409–418.
Hugouvieux-Cotte-Pattat, N., Reverchon, S., Nasser, W., Condemine, G., and Robert-Baudouy, J., 1996, Regulation of pectinase biosynthesis in Erwinia chrysanthemi, in Visser, J. and Voragens, A.G.J., eds., Progress in biotechnology 14—Pectins and pectinases: Amsterdam, Elsevier, p. 311–330.
Hunter, K.A. and Lee, K.C., 1986, Polarographic study of the interaction between humic acids and other surface-active organics in river waters: Water Research, v. 20, p. 1489–1491.
Ishige, T., Tani, A., Sakai, Y., and Kato, N., 2003, Wax ester production by bacteria: Current Opinions in Microbiology, v. 6, p. 244–250.
Jenkinson, D.S., and Ladd, J.N., 1981, Microbial biomass in soil: Measurement and turnover, in Paul, E.A. and Ladd, J.N., Soil Biochemistry: New York, Marcel Dekker, v. 5, p. 415–471.
Joschek, H.-I. And Miller, S.I., 1966, Photooxidation of phenol, cresols, and dihydroxybenzenes: Journal of the American Chemical Society, v. 88, p. 3273–3281.
Jurd, Leonard, 1963, Anthocyanins and related compounds. I. Structural transformations of flavylium salts in acidic solutions: The Journal of Organic Chemistry, v. 28, p. 987–991.
Kähkönen, M.P. and Heinonen, Marina, 2003, Antioxidant activity of anthocyanins and their aglycons: Journal of Agricultural and Food Chemistry, v. 51, p. 628–633.
Kahle, M., Kleber, M., Torn, M.S., and Jahn, R., 2003, Carbon storage in coarse and fine clay fractions of illitic soils: Soil Science Society of America Journal, v. 61, p. 1732–1739.
Karapanagiotti, H.K., Kleineidam, Sybille; Sabatini, D.A., Grathwohl, Peter; and Ligouis, Bertrand, 2000, Impacts of heterogeneous organic matter on phenanthrene sorption, Equilibrium and kinetic studies with aquifer material: Environmental Science and Technology, v. 34, p. 406–414.
Knicker, Heike, 2002, The feasibility of using DCPMAS 15N 13C NMR spectroscopy for a better characterization of immobilized 15N during incubation of 13C- and 15N-enriched plant material: Organic Geochemistry, v. 33, p. 237–246.
Knicker, Heike; Lüdemann, H.-D. and Haider, K., 1997, Incorporation studies of NH4+ during incubation of organic residues by 15N-CPMAS-NMR-spectroscopy: European Journal of Soil Science, v. 48, p. 431–441.
Kolattukudy, P.E., 2001, Polyesters in higher plants: Advances in Biochemical Engineering/Biotechnology, v. 71, p. 1–49.
Kolattukudy, P.E. and Espelie, K.E., 1989, Chemistry, biochemistry, and function of suberin and associated waxes, in Rowe, J.W., ed., Natural products of woody plants I— Chemicals extraneous to the lignocellulosic cell wall: Berlin, Springer-Verlag, p. 304–367.
Kono, Hiroyuki; Yunoki, Shunji; Shikano, Tamio; Fujiwara, Masashi; Erata, Tomoki; and Takai, Mitsuo, 2002, CP/MAS 13C NMR study of cellulose and cellulose derivatives. 1. Complete assignment of the CP/MAS 13C NMR spectrum of the native cellulose: Journal of the American Chemical Society, v. 124, p. 7506–7511.
Kononova, M.M., 1966, Soil organic matter—Its nature, its role in soil formation and in soil fertility: Oxford, U.K., Pergamon Press, 544 p.
Laird, D.A., Martens, D.A., and Kingery, W.L., 2001, Nature of clay-humic complexes in agricultural soil: I. Chemical biochemical and spectroscopic analyses: Soil Science Society of America Journal, v. 65, p. 1413–1418.
Lam, T.B.T., Iiyama, Kenji; and Stone, B.A., 1992, Cinnamic acid bridges between cell wall polymers in wheat and phalaris internodes: Phytochemistry, v. 31, p. 1179–1183.
Larson, R.A. and Marley, K.A., 1999, Singlet oxygen in the environment in Boule, P., ed., The handbook of environmental chemistry, v. 2 part L, Environmental photochemistry: Berlin, Springer-Verlag, p. 123–137.
Larsson, P.T., Hult, Eva-Lena; Wickholm, Kristina; Pettersson, Erik; and Iversen, Tommy, 1999, CP/MAS 13C-NMR spectroscopy applied to structure and interaction studies on cellulose I: Solid State Nuclear Magnetic Resonance, v. 15, p. 31–40.
Lawoko, Martin; Henriksson, Gunnar; and Gellerstedt, Gören, 2003, New method for quantitative preparation of lignin-carbohydrate complex from unbleached softwood kraft pulp, Lignin-polysaccharide networks I: Holzforschung, v. 57, p. 69–74.
Ledl, Franz and Schleicher, Erwin, 1990, New aspects of the Maillard reaction in foods and in the human body: Angewandte Chemie International Edition in English, v. 29, p. 565–594.
Leenheer, J.A., Croué, Jean-Philippe; Benjamin, Mark; Korshin, G.V., Hwang, C.J., Bruchet, Auguste; and Aiken, G.R., 2000, Comprehensive isolation of natural organic matter from water for spectral characterization and reactivity testing, in Barrett, S.E., Krasner, S.W., and Amy, G.L., eds., Natural organic matter and disinfection by-products—Characterization and control in drinking water: Washington, D.C., American Chemical Society Symposium Series 761, p. 68–83.
Leenheer, J.A., Nanny, M.A., and McIntire, Cameron, 2003, Terpenoids as major precursors of dissolved organic matter in landfill leachates, surface water and groundwater: Environmental Science and Technology, v. 37, p. 2323–2331.
Lehmann, Johannes; da Silva, J.P., Jr., Steiner, Christoph; Nehls, Thomas; Zech, Wolfgang; and Glaser, Bruno, 2003, Nutrient availability and leaching in an archaeological Anthrosol and a Ferralsol of the Central Amazon basin—Fertilizer, manure and charcoal amendments: Plant and Soil, v. 249, p. 343–357.
Leshem, Y.Y., 1988, Plant senescent processes and free radicals: Free Radical Biology & Medicine, v. 5, p. 39–49.
Lichtfouse, Éric; Chenu, Claire; Baudin, François; Leblond, Claudette; Da Silva, Martine; Behar, Françoise; Derenne, Sylvie; Largeau, Claude; Wehrung, Patrick; and Albrecht, Pierre, 1998, A novel pathway of soil organic matter formation by selective preservation of resistant straight-chain biopolymers, Chemical and isotope evidence: Organic Geochemistry, v. 28, p. 411–415.
Limbeck, Andreas; Kulmala, Markku; and Puxbaum, Hans, 2003, Secondary organic aerosol formation in the atmosphere via heterogeneous reaction of gaseous isoprene on acidic particles: Geophysical Research Letters, v. 30(19), 1996, doi:10.1029/2003GL017738.
Lou, Xianwen; Zhu, Qingshan; Lei, Ze; van Dongen, J.L.J., and Meijer, E.W., 2004, Simulation of size exclusion chromatography for characterization of supramolecular complex, A theoretical study: Journal of Chromatography A, v. 1029, p. 67–75.
Mäder, Paul; Fliebach, Andreas; Dubois, David; Gunst, Lucie; Fried, Padruot; and Niggli, Urs, 2002, Soil fertility and biodiversity in organic farming: Science, v. 296, p. 1694–1697.
Mariani, C., and Wolters-Arts, M., 2000, Complex waxes: The Plant Cell, v. 12, 1795–1798.
Martens, D.A., 2001, Nitrogen cycling under different soil management systems: Advances in Agronomy, v. 70, p. 143–192.
Martens, D.A. and Loeffelmann, K.L., 2003, Soil amino acid composition quantified by acid hydrolysis and anion chromatography-pulsed amperometry: Journal of Agricultural and Food Science, v. 51, p. 6521–6529.
Martin, V.J.J., Yu, Zhongtang; and Mohn, W.W., 1999, Recent advances in understanding resin acid biodegradation, Microbial diversity and metabolism: Archives of Microbiology, v. 172, p. 131–138.
Masiello, C.A. and Druffel, E.R.M., 1998, Black carbon in deep-sea sediments: Science, v. 280, p. 1911–1913.
McFall, Elizabeth and Newman, E.B., 1996, Amino acids as carbon sources, in Neidhardt, F.C., Curtiss, Roy III; Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, Boris; Reznikoff, W.S., Riley, Monica; Schaechter, Moselio; Umbarger, H.E., eds., Escherichia coli and salmonella—Cellular and molecular biology, 2nd edition, v. 1: Washington, D.C., American Society for Microbiology Press, p. 358–379.
McGrath, T.E., Chan, W.G. and Hajaligol, M.R., 2003, Low temperature mechanism for the formation of polycyclic aromatic hydrocarbons from pyrolysis of cellulose: Journal of Analytical and Applied Pyrolysis, v. 66, p. 51–70.
McIntosh, T.J., Pollastri, M.P., Porter, N.A., and Simon, S.A., 1999, Polyphenols increase adhesion between lipid bilayers by forming interbilayer bridges, in Gross, G.G., Hemingway, R.W., Yoshida, Takashi; and Branham, S.J., Plant polyphenols 2—Chemistry, biology, pharmacology, ecology: New York, Kluwer Academic/Plenum Publishers, p. 451–470.
McKenna, F., El-Tarabily, K.A., Petrie, S., Chen, C., and Dell, B., 2002, Application of actinomycetes to soil to ameliorate water repellency: Applied Microbiology, v. 35, p. 107–112.
Merdy, Patricia; Guillon, Emmanuel; Aplincourt, Michel; and Duomonceau, Jacques, 2000, Interaction of metallic cations with lignins. Part 1, Stability of iron (III), manganese (II) and copper (II) complexes with phenolic lignin model compounds—Coumaric, ferulic and sinapic acids and coniferyl alcohol: Journal of Chemical Research (S), 2000, p. 76–77.
Mohamed, M.E-S., Zaar, Annette; Ebenau-Jehle, Christa; and Fuchs, Georg, 2001, Reinvestigation of a new type of aerobic benzoate metabolism in the proteobacterium Azoarcus evansii: Journal of Bacteriology, v. 183, p. 1899–1908.
Mohnen, Debra, 1999, Biosynthesis of pectins and galactomannans, in Pinto, B.M., ed., Comprehensive natural products chemistry, v. 3, Carbohydrates and their derivatives including tannins, cellulose, and related lignins: Amsterdam, Elsevier, p. 497–527.
Neidhardt, F.C., and Umbarger, H.E., 1996, Chemical composition of Escherichia coli, in Neidhardt, F.C., Curtiss, Roy III; Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, Boris; Reznikoff, W.S., Riley, Monica; Schaechter, Moselio; Umbarger, H.E., eds., Escherichia coli and salmonella—Cellular and molecular biology, 2nd edition, v. 1: Washington, D.C., American Society for Microbiology Press, p. 13–16.
Nierop, K.G.J. and Buurman, P., 1999, Water-soluble organic matter in incipient podzols, Accumulation in B horizons or in fibres?: European Journal of Soil Science, v. 50, p. 701–711.
Nierop, K.G.J., van Lagen, Barend; and Buurman, Peter, 2001, Composition of plant tissues and soil organic matter in the first stages of vegetation succession: Geoderma, v. 100, p. 1–24.
Nimlos, M.R., Blanksby, S.J., Ellison, G.B. and Evans, R.J., 2003, Enhancement of 1,2-dehydration of alcohols by alkali cations and protons, A model for dehydration of carbohydrates: Journal of Analytical and Applied Pyrolysis, v. 66, p. 3–27.
Ohta, Y., 1989a, Simple organic acids, in Rowe, J.W., ed., Natural products of woody plants I—Chemicals extraneous to the lignocellulosic cell wall: Berlin, Springer-Verlag, p. 259–274.
Ohta, Y., 1989b, Complex aliphatic and alicyclic extractives, in Rowe, J.W., ed., Natural products of woody plants I— Chemicals extraneous to the lignocellulosic cell wall: Berlin, Springer-Verlag, p. 274-299.
Ollis, D.F., 1985, Contaminant degradation in water: Environmental Science and Technology, v. 19, p. 480–484.
Ollis, D.F., Pelizzetti, Ezio, and Serpone, Nick, 1991, Destruction of water contaminants: Environmental Science and Technology, v. 25, p. 1523–1529.
Oros, D.R., Mazurek, M.A., Baham, J.E., and Simoneit, B.R.T., 2002, Organic tracers from wild fire residues in soils and rain/river wash-out: Water, Air, and Soil Pollution, v. 137, 203–233.
Osburn, C.L., Morris, D.P., Thorn, K.A., and Moeller, R.E., 2001, Chemical and optical changes in freshwater dissolved organic matter exposed to solar radiation: Biogeochemistry, v. 54, p. 251-278.
Pacheco, M.L., Peña-Méndez, E.M., and Havel, J., 2003, Supramolecular interactions of humic acids with organic and inorganic xenobiotics studied by capillary electrophoresis: Chemosphere, v. 51, p. 95–108.
Pagni, R.M., and Sigman, M.E., 1999, The photochemistry of PAHs and PCBs in water and on solids in Boule, P., ed., The handbook of environmental chemistry, v. 2 part L, Environmental photochemistry: Berlin, Springer-Verlag, p. 139–179.
Parameswaran, N. and Wilhelm, G.E., 1979, Micromorphology of naturally degraded beech and spruce barks: European Journal of Forest Pathology, v. 9, p. 103–112.
Pérez, J., Muñoz-Dorado, J., de la Rubia, T., and Martínez, J., 2002, Biodegradation and biological treatments of cellulose, hemicellulose and lignin, An overview: International Microbiology, v. 5, p. 53–63.
Philosoph-Hadas, Sonia; Meir, Shimon; Akiri, Bezalel; and Kanner, Joseph, 1994, Oxidative defense systems in leaves of three edible herb species in relation to their senescence rates: Journal of Agricultural and Food Chemistry, v. 42, p. 2376–2381.
Piccolo, Alessandro, 2001, The supramolecular structure of humic substances: Soil Science, v. 166, p. 810–832.
Pimentel, David; Harvey, C., Resosudarmo, P., Sinclair, K., Kurz, D., McNair, M., Crist, S., Shpritz, L., Fitton, L., Saffouri, R., and Blair, R., 1995, Environmental and economic costs of soil erosion and conservation benefits: Science, v. 267, p. 1117–1123.
Pine, S.H., Hendrickson, J.B., Cram, D.J., Hammond, G.S., 1980, Organic Chemistry (4th ed): New York, McGraw-Hill Book Company, p. 966–1000.
Preston, C.M., Shaw, C.H., Bhatti, J.S., and Siltanen, R.M., 2000, Soil C and N pools in forested upland and non-forested lowland sites along the boreal forest transect case study in Central Canada, in Shaw, C.H. and Apps, M.J., eds., The role of boreal forests and forestry in the global carbon budget, Proceedings of the International Boreal Forest Research Association 2000 Conference, May 8–12, 2000, Edmonton, Alberta, Canada: Edmonton, Natural Resources Canada, Canadian Forest Service, p. 155–178.
Preston, C.M., Troyfymow, J.A., Niu, J., and Fyfe, C.A., 1998, 13CPMAS-NMR spectroscopy and chemical analysis of coarse woody debris in coastal forest of Vancouver Island: Forest Ecology and Management, v. 111, p. 51–68.
Qualls, R.G. and Haines, B.L., 1992, Biodegradability of dissolved organic matter in forest throughfall, soil solution, and stream water: Soil Science Society of America Journal, v. 56, p. 578–586.
Qualls, R.G., Takiyama, Akiko; and Wershaw, R.L., 2003, Formation and loss of humic substances during decomposition in a pine forest floor: Soil Science Society of America Journal, v. 67, p. 899–909.
Quiocho, F.A., 1986, Carbohydrate-binding proteins, Tertiary structures and protein-sugar interactions: Annual Reviews of Biochemistry, v. 55, p. 287–315.
Ralph, John; Lapierre, Catherine; Marita, J.M., Kim, Hoon; Lu, Fachuang; Hatfield, R.D., Ralph, Sally; Chapple, Clint; Franke, Rochus; Hemm, M.R., Van Doorsselaere, Jan; Sederoff, R.R., O'Malley, D.M., Scott, J.T., MacKay, J.J., Yahiaoui, Nabila; Boudet, Alain-M., Pean, Michel; Pilate, Gilles; Jouanin, Lise; Boerjan, Wout, 2001, Elucidation of new structures in lignins of CAD- and COMT-deficient plants by NMR: Phytochemistry, v. 57, p. 993–1003.
Reitzer, L.J., 1996a, Sources of nitrogen and their utilization, in Neidhardt, F.C., Curtiss, Roy III; Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, Boris; Reznikoff, W.S., Riley, Monica; Schaechter, Moselio; Umbarger, H.E., eds., Escherichia coli and salmonella—Cellular and molecular biology, 2nd edition, v. 1: Washington, D.C., American Society for Microbiology Press, p. 380–390.
Reitzer, L.J., 1996b, Ammonia assimilation and the biosynthesis of glutamine, glutamate, aspartate, asparagines, L-alanine and D-alanine, in Neidhardt, F.C., Curtiss, Roy III; Ingraham, J.L., Lin, E.C.C., Low, K.B., Magasanik, Boris; Reznikoff, W.S., Riley, Monica; Schaechter, Moselio; Umbarger, H.E., eds., Escherichia coli and salmonella—Cellular and molecular biology, 2nd edition, v. 1: Washington, D.C., American Society for Microbiology Press, p. 391–407.
Richard, Claire, and Grabner, Gottfried, 1999, Mechanism of phototransformation of phenol and derivatives in aqueous solution in Boule, P., ed., The handbook of environmental chemistry, v. 2, part L, Environmental photochemistry: Berlin, Springer-Verlag, p. 217–240.
Rogge, W.F., Hildemann, L.M., Mazurek, M.A., Cass, G.R., and Simoneit, B.R.T., 1998, Sources of fine organic aerosol. 9. Pine, oak and synthetic log combustion in residential fireplaces: Environmental Science and Technology, v. 32, p. 13–22.
Rontani, Jean-François; Mouzdahir, Abdelkrim; Michotey, Valerie; Caumette, Pierre; and Bonin, Patricia, 2003, Production of polyunsaturated isoprenoid wax ester during aerobic metabolism of squalene by Marinobacter squalenivorans sp. nov.: Applied and Environmental Microbiology, v. 69, p. 4167–4176.
Saake, Bodo; Argyropoulos, D.S., Beinhoff, Ottokar; and Faix, Oskar, 1996, A comparison of lignin polymer models (DHPs) and lignins by 31P NMR spectroscopy: Phytochemistry, v. 43, p. 499–507.
Salas, Erika; Fulcrand, Hélène; Meudec, Emmanuelle; and Cheynier, Véronique, 2003, Reactions of anthocyanins and tannins in model solutions: Journal of Agricultural and Food Chemistry, v. 51, p. 7951–7961.
Sanders, E.B., Goldsmith, A.I, and Seeman, J.I., 2003, A model that distinguishes the pyrolysis of D-glucose, D-fructose, and sucrose from that of cellulose. Application to the understanding of cigarette smoke formation: Journal of Analytical and Applied Pyrolysis, v. 66, p. 29–50.
Scalbert, Augustin, 1991, Antimicrobial properties of tannins: Phytochemistry, v. 30, p. 3875–3883.
Schmidt, M.W.I., Skjemstad, J.O., Gehrt, E., and Kögel-Knabner, Ingrid, 1999, Charred organic carbon in German chernozemic soils: European Journal of Soil Science, v. 50, p. 351–365.
Schmidt, M.W.I., and Kögel-Knabner, Ingrid, 2002, Organic matter in particle-size fractions from A and B horizons of a Haplic Alisol: European Journal of Soil Science, v. 53, p. 383–391.
Schmidt, M.W.I., Masiello, C.A., and Skjemstad, J.O., 2003, Final recommendations for reference materials in black carbon analysis: EOS, Transactions, American Geophysical Union, v. 84, p. 582.
Schulten, H.-R. and Schnitzer, Morris, 1995, Three-dimensional models for humic acids and soil organic matter: Naturwissenschaften, v. 82, p. 487–498.
Sein, L.A., Jr., Varnum, J.M., and Jansen, S.A., 1999, Conformational modeling of a new building block of humic acid, Approaches to the lowest energy conformer: Environmental Science and Technology, v. 33, p. 546–552.
Shafizadeh, Fred, 1982, Introduction to pyrolysis of biomass: Journal of Analytical and Applied Pyrolysis, v. 3, p. 283–305.
Shafizadeh, Fred, 1984, The chemistry of pyrolysis and combustion in Rowell, Roger, ed., The chemistry of solid wood: Washington, American Chemical Society Advances in Chemistry Series 207, p. 489–529.
Sharma, R.K., and Hajaligol, M.R., 2003, Effect of pyrolysis conditions on the formation of polycylic aromatic hydrocarbons (PAHs) from polyphenolic compounds: Journal of Analytical and Applied Pyrolysis, v. 66, p. 123–144.
Sharma, R.K., Chan, W.G., Seeman, J.I., and Hajaligol, M.R., 2003, Formation of low molecular weight heterocycles and polycyclic aromatic compounds (PACs) in the pyrolysis of ∝-amino acids: Journal of Analytical and Applied Pyrolysis, v. 66, p. 97–121.
Simoneit, B.R.T., 2002, Biomass burning—a review of organic tracers for smoke from incomplete combustion: Applied Geochemistry, v. 17, p. 129–162.
Simoneit, B.R.T., Rogge, W.F., Mazurak, M.A., Standley, L.J., Hildemann, L.M., and Cass, G.R., 1993, Lignin pyrolysis products, lignans, and resin acids as specific tracers of plant classes in emissions from biomass combustion: Environmental Science and Technology, v. 27, p. 2533–2541.
Simpson, A.J., Kingery, W.L., Hayes, M.H.B., Spraul, Manfred; Humpfer, Eberhard; Dvortsak, Peter; Kerssebaum, Rainer; Godejohann, Markus; and Hofmann, Martin, 2002, Molecular structures and associations of humic substances in the terrestrial environment: Naturwissenschaften, v. 89, p. 84–88.
Simpson, David; Winiwarter, Wilfried; Börjesson, Gunnar; Cinderley, Steve; Ferreiro, Antonio; Guenther, Alex; Hewitt, C.N., Janson, Robert; Khalil, M.A.K., Owen, Susan; Pierce, T.E., Puxbaum, Hans; Shearer, Martha; Skiba, Ute; Steinbrecher, Rainer; Tarrasón, Leonor; Öquist, M.G., 1999, Inventorying emissions from nature in Europe: Journal of Geophysical Research, v. 104(D7), p. 8113–8152.
Six, J., Guggenberger, G., Paustian, K., Haumaier, L., Elliott, E.T., and Zech, W., 2001, Sources and composition of soil organic matter fractions between and within soil aggregates: European Journal of Soil Science, v. 52, p. 607–618.
Skjemstad, J.O., Clarke, P., Taylor, J.A., Oades, J.M., and McClure, S.G., 1996, The chemistry and nature of protected carbon in soil: Australian Journal of Soil Research, v. 32, p. 251–276.
Skjemstad, J.O., Reicosky, D.C., Wilts, A.R., and McGowan, J.A., 2002, Charcoal carbon in U.S. agricultural soils: Soil Science Society of America Journal, v. 66, p. 1249–1255.
Smernik, R.J., and Oades, J.M., 2002, Paramagnetic effects on solid state carbon-13 nuclear magnetic resonance spectra of soil organic matter: Journal of Environmental Quality, v. 31, p. 414–420.
Sohi, S.P., Mahieu, Nathalie; Arah, J.R.M., Powlson, D.S., Madari, Beáta; and Gaunt, J.L., 2001, A procedure for isolating soil organic matter fractions suitable for modeling: Soil Science Society of America Journal, v. 65, p. 1121–1128.
Sojo, M.M., Nuñez-Delicado, Estrella; García-Carmona, Francisco; and Sánchez-Ferrer, Alvaro, 1999, Cyclodextrins as activator and inhibitor of latent banana pulp polyphenol oxidase: Journal of Agricultural and Food Chemistry, v. 47, p. 518–523.
Sposito, Garrison; Skipper, N.T., Sutton, Rebecca; Park, Sung-Ho; Soper, A.K., and Greathouse, J.A., 1999, Surface geochemistry of clay minerals: Proceedings of the National Academy of Sciences USA, v. 96, p. 3358–3364.
Stevenson, F.J., 1994, Humus chemistry—Genesis, composition, reactions (2nd ed.): New York, John Wiley, 496 p.
Sun, RunCang; Fang, J.M., and Tomkinson, J., 2000, Characterization and esterification of hemicelluloses from rye straw: Journal of Agricultural and Food Chemistry, v. 48, p. 1247–1252.
Tauer, Andreas; Elss, Sandra; Frischmann, Matthias; Tellez, Patricia; and Pischetsrieder, Monika, 2004, Influence of thermally processed carbohydrate/amino acid mixtures on the fermentation by Saccharomyces cerevisiae: Journal of Agricultural and Food Chemistry, v. 52, p. 2042–2046.
Terashima, Noritsugu; Hafrén, Jonas; Westermark, Ulla; and VanderHart, D.L., 2002, Nondestructive analysis of lignin structure by NMR spectroscopy of specifically 13C-enriched lignins, Part 1. Solid state study of ginko wood: Holzforschung, v. 56, 43–50.
Tilman, David, 1999, Global environmental impacts of agricultural expansion—The need for sustainable and efficient practices: Proceedings of the National Academy of Sciences, v. 96, p. 5995–6000.
Ulmer, D.C., Leisola, M.S.A., Schmidt, B.H., and Fiechter, Armin, 1983, Rapid degradation of isolated lignins by Phanerochaete chrysosporium: Applied and Environmental Microbiology, v. 45, p. 1795–1801.
van Boekel, M.A.J.S., 2001, Kinetic aspects of the Maillard reaction, A critical review: Nahrung/Food, v. 45, p. 150–159.
van der Werf, M.J., Swarts, H.J., and de Bont, J.A.M., 1999, Rhodococcus erythropolis DCL14 contains a novel degradation pathway for limonene: Applied and Environmental Microbiology, v. 65, p. 2092–2102.
van Oss, C.J., 2003, Long-range and short-range mechanisms of hydrophobic attraction and hydrophilic repulsion in specific and aspecific interactions: Journal of Molecular Recognition, v. 16, p. 177–190.
Vaughan, P.P. and Blough, N.V., 1998, Photochemical formation of hydroxyl radicals by constituents of natural waters: Environmental Science and Technology, v. 32, p. 2947–2953.
Wagner, Karl-Heinz and Elmadfa, Ibrahim, 2003, Biological relevance of terpenoids: Annals of Nutrition and Metabolism, v. 47, p. 95–106.
Wershaw, R.L., 1986, A new model for humic materials and their interactions with hydrophobic chemicals in soil-water or sediment-water systems: Journal of Contaminant Hydrology, v. 1, p. 29–45.
Wershaw, R.L., 1994, Membrane-micelle model for humus in soils and sediments and its relation to humification: U.S. Geological Survey Water-Supply Paper 2410, 48 p.
Wershaw, R.L., 2000, The study of humic substances—In search of a paradigm, in Ghabbour, E.A., and Davies, Geoffrey, eds., Humic substances—Versatile components of plants, soils and water: Cambridge, Royal Society of Chemistry, p. 1–7.
Wershaw, R.L. and Hayes, T.M., 2001, Solubilization of anthropogenic compounds by humic substances, in Clapp, C.E., Hayes, M.H.B., Senesi, N., Bloom, P.R., and Jardine, P.M., eds., Humic substances and chemical contaminants: Madison, Wisconsin, Soil Science Society of America, p. 165–176.
Wershaw, R.L. and Kennedy, K.R., 1998, Use of 13C NMR and FTIR for elucidation of degradation pathways during natural litter decomposition and composting. IV. Characterization of humic and fulvic acids extracted from senescent leaves, in Davies, Geoffrey, and Ghabbour, E.A., eds., Humic substances—Structures, properties and uses: Cambridge, Royal Society of Chemistry, p. 60–68.
Wershaw, R.L., Kennedy, K.R., and Henrich, J.E., 1998a, Use of 13C NMR and FTIR for elucidation of degradation pathways during natural litter decomposition and composting. II. Changes in leaf composition after senescence, in Davies, Geoffrey, and Ghabbour, E.A., eds., Humic substances—Structures, properties and uses: Cambridge, Royal Society of Chemistry, p. 29–46.
Wershaw, R.L., Leenheer, J.A., and Kennedy, K.R., 1998b, Use of 13C NMR and FTIR for elucidation of degradation pathways during natural litter decomposition and composting. III. Characterization of leachate from different types of leaves, in Davies, Geoffrey, and Ghabbour, E.A., eds., Humic substances—Structures, properties and uses: Cambridge, Royal Society of Chemistry, p. 47–60.
Wershaw, R.L., Llaguno, E.C., and Leenheer, J.A., 1996, Mechanism of formation of humus coatings on mineral surfaces, 3. Composition of adsorbed organic acids from compost leachate on alumina by solid-state 13C NMR: Colloids and Surfaces A, v. 108, p. 213–223.
Wershaw, R.L. and Pinckney, D.J., 1973, The fractionation of humic acids from natural water systems: Journal of Research of the U.S. Geological Survey, v. 1, p. 361–366.
Wershaw, R.L. and Pinckney, D.J., 1980, Isolation and characterization of clay-humic complexes, in Baker, R.A., ed., Contaminants and sediments, v. 2—Analysis, chemistry and biology: Ann Arbor, Ann Arbor Science Publishers, p. 207–219.
Wershaw, R.L., Pinckney, D.J., Llaguno, E.C., and Vincente-Beckett, Victoria, 1990, NMR characterization of humic acid fractions from different Philippine soils and sediments: Analytica Chemica Acta, v. 232, p. 31–42.
Wershaw, R.L., Rutherford, D.W., Leenheer, J.A., Kennedy, K.R., Cox, L.G., and Koci, D.R., 2003, Biogeochemical processes that produce dissolved organic matter from wheat straw: U.S. Geological Survey Water-Resources Investigations Report 03-4213, persistent URL https://pubs.water.usgs.gov/wri034213.
Wershaw, R.L., Thorn, K.A., and Pinckney, D.J., 1988, Characterization of humic acid fractions by C13 nuclear magnetic resonance spectroscopy: Environmental Technology Letter, v. 9, p. 53–62.
Xing, Baoshan, 1998, Nonlinearity and competitive sorption of hydrophobic organic compounds in humic substances, in Davies, Geoffrey, and Ghabbour, E.A., eds., Humic substances—Structures, properties and uses: Cambridge, Royal Society of Chemistry, p. 173–183.
Yang, Yaning and Sheng, Guangyao, 2003, Enhanced pesticide sorption by soils containing particulate matter from crop residue burns: Environmental Science and Technology, v. 37, p. 3635–3639.
Yu, Zhongtang; Stewart, G.R., and Mohn, W.W., 2000, Apparent contradiction, Psychrotolerant bacteria from hydrocarbon-contaminated arctic tundra soils that degrade diterpenoids synthesized by trees: Applied and Environmental Microbiology, v. 66, p. 5148–5154.
Zaar, Annette; Eisenreich, Wolfgang; Bacher, Adelbert; and Fuchs, Georg, 2001, A novel pathway of aerobic benzoate catabolism in the bacteria Azoarcus evansii and Bacillus stearothermophilus: The Journal of Biological Chemistry, v. 276, p. 24997–25004.
Zafiriou, O.C., Joussot-Dubien, Jacques; Zepp, R.G., and Zika, R.G., 1984, Photochemistry of natural waters: Environmental Science and Technology, v. 18, p. 358A–371A.
Zang, Xu; van Heemst, J.D.H., Dria, K.J., and Hatcher, P.G., 2000, Encapsulation of protein in humic acid from a histosol as an explanation for the occurrence of organic nitrogen in soil and sediment: Organic Geochemistry, v. 31, p. 679–695.
Zang, Xu; Nguyen, R.T., Harvey, H.R., Knicker, Heike; and Hatcher, P.G., 2001, Preservation of proteinaceous material during the degradation of the green alga Botryococcus braunii, A solid-state 2D 15N 13C NMR spectroscopy study: Geochimica et Cosmochimica Acta, v. 65, p. 3299–3305.
Zepp, R.G., and Schlotzhauer, P.F., 1983, Influence of algae on photolysis rates of chemicals in water: Environmental Science and Technology, v. 17, p. 462–468.
Zepp, R.G., Callaghan, T.V., and Erickson, D.J., 1995, Effects of increased solar ultraviolet radiation on biogeochemical cycles: Ambio, v. 24, p. 181–187.
Zimmels, Y., and Metzer, A., 1976, Influence of association properties of sodium oleate in aqueous solution on solubility of parafinic gases: Journal of Colloid and Interface Science, v. 57, p. 75–84.
Zinkel, D.F., 1989, Fats and fatty acids, in Rowe, J.W., ed., Natural products of woody plants, I—Chemicals extraneous to the lignocellulosic cell wall: Berlin, Springer-Verlag, p. 299–304.
Document Accessibility: Adobe Systems Incorporated has information about PDFs and the visually impaired. This information provides tools to help make PDF files accessible. These tools convert Adobe PDF documents into HTML or ASCII text, which then can be read by a number of common screen-reading programs that synthesize text as audible speech. In addition, an accessible version of Acrobat Reader 6.0, which contains support for screen readers, is available. These tools and the accessible reader may be obtained free from Adobe at Adobe Access.
| AccessibilityFOIAPrivacyPolicies and Notices | |
 |
|