
Characterization of dissolved and particulate natural organic matter
Figure 1. Diagram of isolation and fractionation procedure.
Figure 1. Diagram of isolation and fractionation procedure—Continued.
Figure 2a. Infrared spectra of particulate fractions.
Figure 2b. Infrared spectra of dissolved fractions.
Figure 2c. Infrared spectra of HPO-A sub-fractions.
Figure 3a. Solid-state 13C nuclear magnetic resonance spectra of particulat...
Figure 3b. Solid-state 13C nuclear magnetic resonance spectra of dissolved ...
Figure 3c. Solid-state 13C nuclear magnetic resonance spectra of HPO-A sub-...
Table 1. Results of whole-water analyses.
Table 2. Yields and organic carbon contents of the fractions
Table 3. The carbon chemical shifts of types of carbon atoms commonly foun...
Table 4. Elemental analyses of fractions on an as received basis
Table 5. Amino acid composition of Neversink Reservoir natural organic matter.
Table 7. Relative concentrations of carbohydrates in Neversink fractions
Natural organic matter (NOM) was isolated from the water of the Neversink Reservoir, part of the New York City water supply, located in the Catskill Mountains of New York. The NOM was fractionated into the following nine different fractions by the isolation procedure: (1) coarse particulates, (2) fine-particulate organics, (3) solvent-extractable organics, (4) hydrophobic neutrals (HPON fraction), (5) dissolved colloids, (6) bases, (7) hydrophobic acids (HPOA), (8) transphilic acids + neutrals (TPI-A+N), and (9) hydrophilic acids + neutrals (HPI-A+N). Each of these fractions, with exception of the first and the third which were too small for the complete series of analyses, was characterized by elemental, carbohydrate, and amino acid analyses, and by nuclear magnetic resonance and infrared spectrometry. The data obtained from these analyses indicate (1) that the fine-particulate organics and colloids are mainly composed of peptidoglycans, and lipopolysaccharides derived from algal, bacterial, and fungal cell walls, (2) that the HPO-N fraction most likely consists of a mixture of alicyclic terpenes and carbohydrates, (3) that the HPOA fraction consists mainly of lignin components conjugated to carbohydrates, (4) that the TPI-A+N and the HPI-A+N fractions most likely represent complex mixtures of relatively low molecular weight carboxylic acids derived from terpenes, carbohydrates, and peptides, and (5) that the base fraction is composed of free amino acids, browning reaction products, and peptide fragments.
The organic compounds in surface waters and ground waters arise from natural and athropogenic sources. Natural organic matter (NOM) is either formed in place (autochthonous) or is formed outside of the water body and then transported into the water body (allochthonous). The composition of allochthonous NOM in a given surface-water body is dependent on both on the composition of the plant-derived precursor compounds and natural diagenetic processes that alter the composition of the mixture of precursors (Wershaw, 2004). As plant organs (for example, leaves) degrade in natural systems, soluble organic compounds are leached from the tissue. These organic compounds are the precursors of NOM. Leenheer and others (2003, 2004) and Wershaw and others (1998, 2003) demonstrated that the chemical composition of NOM is reflective of the chemical components of the plant tissue from which it is derived. For example, Wershaw and others (2003) found that NOM derived mainly from decomposing wheat straw is composed of hemicellulose chains cross-linked to lignin oligomers. Similar structures exist in the hemicellose matrix of wheat straw cell walls. Autochthonous NOM is generally formed by micro-organisms living in a water body.
Diagenetic processes that take place in soils and aquifers alter the composition of NOM. Sorption on mineral surfaces and microbial degradation are the most common of these processes. Wershaw and others (1995; 1996a,b) studied the sorption of NOM from compost leachate on alumina surfaces. They found that organic acids in NOM are strongly adsorbed by alumina, and that those NOM components that form multidentate complexes with ligands on alumina surfaces are preferentially adsorbed. Micro-organisms metabolize plant-derived NOM and produce new types of compounds; for example, Leenheer and others (2004) showed that much of the colloidal fraction of NOM in the Great Salt Lake is composed of amino sugar components derived from bacterial and algal cell walls. Lower molecular weight compounds are, no doubt, also produced by micro-organisms.
Leenheer has developed a comprehensive procedure for the fractionation and characterization of NOM (Leenheer and others, 2000; 2004). In this procedure particulate organic matter is separated from dissolved organic matter (DOM), and the DOM is fractionated according to polarity. This procedure, which has been applied so far to only a few natural waters, allows one to isolate and characterize hydrophilic and colloidal fractions that were lost by the procedures that were used previously (Leenheer and others, 2000).
NOM interacts with all of the chemical components of natural waters; these interactions alter the behavior of pollutants in surface and ground waters. For example, the solubilities of hydrophobic anthropogenic compounds are enhanced by NOM (Wershaw and Hayes, 2001), and NOM forms complexes with metal ions that affect the bioavailability and toxicity of the metals to living organisms (Karlsson and others, 2005). Other types of reactions, such as, the hydrolysis of anthropogenic compounds, are probably also affected by NOM. The isolation and characterization of NOM fractions are of particular importance because each NOM fraction has a unique set of properties. For example, Vignati and others (2005) have shown that the toxicity of contaminants in natural waters is altered by interactions with the colloid fraction of NOM.
This report describes a characterization of dissolved and particulate NOM in a water sample collected from Neversink Reservoir in August 2004. The NOM from the Neversink Reservoir water is the first NOM from an oligotrophic lake with minimal anthropogenic input to have been characterized by this procedure. The Neversink Reservoir, located in the Catskill Mountains of New York, is part of the New York City water supply. The geologic and hydrologic setting of the reservoir has been described in detail by Lawrence and others (2001) and Stoddard and Murdoch (1991). The area of the Neversink Reservoir watershed is 166 km2; ninty-five percent of the area consists of hardwood forest. The forest is composed mainly of American beech (Fagus grandifolia), sugar maple (Acer saccharum), yellow birch (Betula alleghaniensis), and balsam fir (Abies balsamea). The forest floor is covered by a porous litter layer of decomposing leaves and branches. The soils in the region are well-drained Inceptisols 0.5 to 1 m thick that have been derived from glacial till. These highly permeable conditions have led Lawrence and others (2001) to conclude that "Most, if not all, precipitation that falls within the watershed infiltrates the soil before reaching stream channels***."
The water sample from Neversink Reservoir was collected on August 10, 2004, from a tap at the inlet to the aqueduct that carries the water to the New York City water system. The water was not treated prior to introduction into the aqueduct. The sample was placed in 20-L (liter) rigid polyethylene jugs. The jugs were placed in insulated cooler chests, and the chests were iced. The chests were shipped overnight to the National Water Quality Laboratory (NWQL) in Denver, Colorado. The chilled water sample containers were received intact and removed from the iced shipping coolers to a refrigeration unit. The sample was kept refrigerated until processed.
Pesticide and water-quality analyses were performed by the National Water Quality Laboratory of the U.S. Geological Survey. The pesticides were analyzed as described by Lindley and others (1996), Madsen and others (2003) and Zaugg and others (1995). The metal analyses were performed as described by American Public Health Association (1998), Faires (1993), Fishman (1993), Fishman and Friedman (1989), Garbarino (1999), Garbarino and Damrau (2001), and McLain (1993); nitrogen and phosphorous species were analyzed as described by Patton and Kryskalla (2003).
The total organic carbon (TOC) concentration of the whole water was measured by persulfate wet oxidation in a gas-tight reaction vessel as described by Aiken (1992). Samples were introduced into the reaction vessel through a fixed-volume sample loop. Prior to oxidation the samples were acidified with phosphoric acid and purged with nitrogen to remove dissolved carbon dioxide.
The basic principles behind the isolation and fractionation procedure are relatively simple. The particulate and colloidal fractions are isolated by filtration and dialysis. The truly dissolved NOM (DOM) is then fractionated according to polarity by sequential sorption chromatography on XAD-8 and XAD-4 resins (Aiken, 1985). XAD-8 resin is an acrylic ester resin that is more hydrophilic than XAD-4 (styrene divinylbenzene resin). The least polar DOM fraction, the hydrophobic neutral fraction (HPON fraction), is sorbed on an XAD-8 resin column without pH adjustment; by reducing the pH of the water that passes through the column a second more polar fraction, the hydrophobic acid fraction (HPOA), is isolated. A third even more polar fraction, the transphilic acids + neutrals fraction (TPIA+N), is isolated by sorption on an XAD-4 column. The most polar fraction, the hydrophilic acids plus neutrals (HPIA+N), is isolated after sorption of the transphilic fraction by a multi-step precipitation procedure, volatilization of some inorganic ions, and ion exchange. The base fraction is isolated on a cation exchange resin.
The entire water sample was first passed through a 63-micrometer (µm) screen into a pressure filter apparatus. Because the material mass retained on the screen was small, the entire bulk sample was passed through the same screen with no interim collection or cleanout. Collected particulates were washed into a receiving flask and lyophilized (freeze-dried). The lyophilized solids were labeled Coarse-Particulate Organic Matter (see fig. 1 for diagram of isolation procedure).
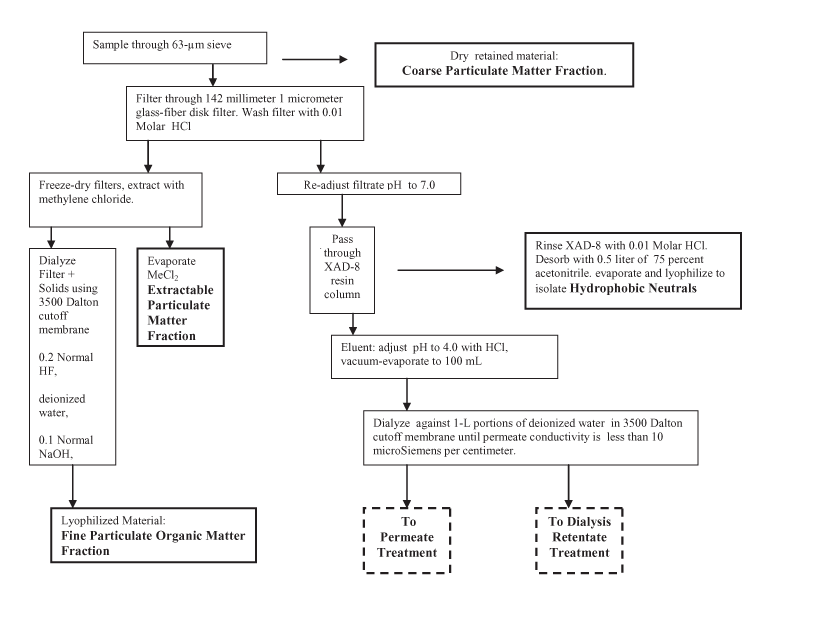
Figure 1. Diagram of isolation and fractionation procedure.
The water was then filtered through a nominal 1-µm glass fiber filter, Gelman® Type A-E. A 142mm Millipore® filter holder was used. Filter pads were replaced at approximately 25 liter (L) intervals due to diminished flow. Fourteen filter pads were used. Filtered samples were returned to cleaned sample containers. Filtrate pH was 6.15 and measured conductivity was 30 microSiemens per centimeter (µS/cm). The filtered sample was collected for XAD-8 column separation described below; the total volume of the sample was 319 L.
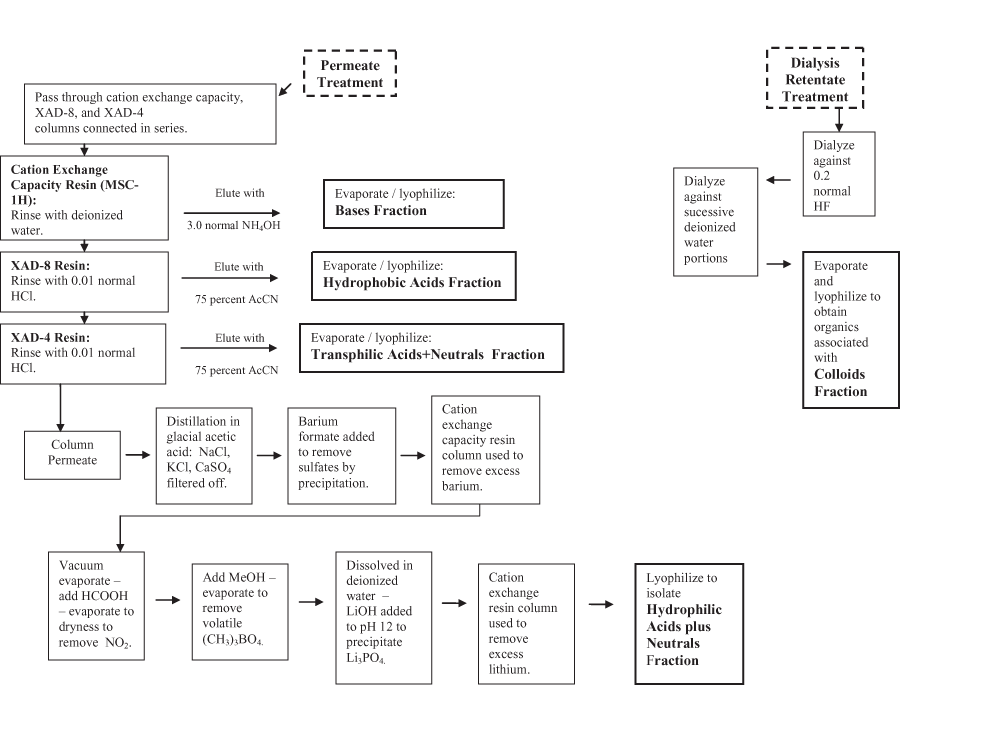
Figure 1. Diagram of isolation and fractionation procedure—Continued.
The loaded filter pads, supported on a 124-mm fritted glass vacuum filter, were washed with pH 2 hydrochloric acid (HCl) solution. The wash solutions were consolidated in a glass bottle, and all of the washed pads were freeze-dried (lyophilized) in a single, large freeze-dry flask. Three and one-half dried filter pads were added to each of four 250-mL teflon jars. The jars were filled with methylene chloride, lidded, and shaken to disaggregate and suspend the glass fiber material and sample. After three days, the methylene chloride solution was separated from the filter pulp and dried. The Extractable Organic Matter Fraction was recovered from methylene chloride.
The four extracted glass fiber filter pulps with remaining particulates were slurried with de-ionized (DI) water into four lengths of Spectra/Por® (Spectrum Laboratories, Inc.) dialysis tubing; 3,500 MWCO (Molecular Weight Cutoff, also designated 3500 Da) porosity tubing was used. Sealed membrane tubes were suspended in 4-L polypropylene beakers containing 0.2N hydrofluoric acid (HF). The HF dialysis solution was changed at two-day intervals until no noticeable glass fiber was left in the membrane tubing (three times). Two dialysis courses in DI water followed the HF treatment to remove excess HF. The DI water solutions were replaced with 0.1N NaOH to remove AlF3 as NaAlO2and NaF from the HF reaction with clays, followed by another DI water dialysis, and finished by dialyzing in pH 2 HCl. The dialyzed sample was lyophilized and labeled Fine-Particulate Organic Matter.
The pH of the entire 319 L of filtered sample was adjusted to 7 and passed through a 1-L bed of Amberlite XAD-8 non-ionic, macroporous resin (Rohm and Haas) packed in a glass column at approximately 15 L per hour. XAD-8 is an acrylic ester, intermediate polarity polymer. The column was prepared by passing an acetonitrile/DI water solution (75 percent/25 percent by volume) followed by a 0.1N NaOH solution. A DI water wash was introduced until the passing solution conductivity dropped to approximately 100 µS/cm. After passing the bulk sample through the column a 0.01N HCl wash was used. The retained organic matter was eluted with an acetonitrile/DI water solution (75 percent/25 percent by volume). The eluent was reduced in volume using a Buechi® Rotavapor R-220 rotary vacuum evaporation unit. The residual eluent was lyophilized and labeled Hydrophobic Neutral Fraction.
The pH of the entire aqueous solution of nonsorbed material was adjusted to 4, and the volume was reduced to approximately 100 mL in a 20-L capacity Buechi® rotary vacuum evaporation unit. This concentrate was introduced into a 3,500 Da dialysis tube and dialyzed against DI water portions in a 1-L graduated cylinder until the dialysis permeate conductivity was approximately 10 µS/cm. All dialysis permeate solutions were combined and saved for subsequent column separation procedure.
The water dialysis retentate was dialyzed against 0.2N HF solutions and further dialyzed against successive DI water batches to remove unreacted HF. The final retentate was evaporated to a small volume and lyophilized to obtain the Dissolved Colloidal Fraction.
The combined dialysis permeate sample (approximately 5 L) described above was passed through three glass columns connected in series packed with (1) cation exchange (CEC) resin (0.5L resin bed CEC, Dowex® MSC-1H), (2) XAD-8 resin (previously described), and (3) Rohm and Haas Amberlite XAD-4 macroporous styrene divinylbenzene resin (0.5L bed volume). Each column was rinsed and eluted with the solutions shown in figure 1. Eluates from individual columns were subsequently evaporated to small volumes and lyophilized to yield specific fractions. The CEC column was rinsed with DI water then eluted with 3N NH4OH; lyophilized residue was labeled Bases Fraction.
The XAD-8 column was rinsed with 0.01N HCl and eluted with an acetonitrile/DI water solution (75 percent/25 percent by volume); the lyophilized residue was labeled Hydrophobic Acids Fraction.
The XAD-4 column was rinsed with 0.01N HCl and eluted with an acetonitrile/DI water solution (75 percent/25 percent by volume); the lyophilized residue was labeled Transphilic Acids + Neutrals Fraction.
The column-pass solution was processed to remove inorganic components as follows. Rotary evaporation was used to reduce the solution volume. Glacial acetic acid was added to the concentrated solution and further evaporated until crystallization of inorganic salts was noted. The salts, notably NaCl, KCl, and CaSO4, were removed by filtration. Barium formate was added to the desalted solution to precipitate any residual SO4=. The solution was filtered to remove BaSO4. The resultant filtrate was passed through the CEC column, used above, to remove excess barium. The column-pass solution was rotary evaporated to a small volume, and formic acid was added and the solution was taken to dryness in the rotary vacuum to remove HNO3 as NO2 gas. Methyl alcohol (MeOH) was added to the dried residue and evaporated to remove borate as volatile (CH3)3BO4. Approximately 100 mL of DI water was added to dissolve the residue. Saturated LiOH solution was added to attain pH 12 to precipitate phosphate as LI3PO4. Residual lithium ions were removed by another pass through the CEC column. The Hydrophilic Acids + Neutrals Fraction was obtained by lyophilization of the resultant treated solution.
The hydrophobic acid fraction was refractionated on a 1-cm diameter column packed with 20 mL of Woelmℜ 100–200 aktiv silica gel (particle size 100–200 µm). The column was washed with 25 mL of water, 50 mL of acetonitrile, and 50 mL of methylene chloride. A solution of 200 mg of the hydrophobic acid fraction dissolved in 3 mL of 75 percent acetonitrile/25 percent water was applied to the column. The acetronitrile-water solution was displaced from the column with 100 mL of methylene chloride. The column was then eluted with 200 mL of ethyl acetate. The ethyl acetate was evaporated to yield the first sub-fraction. After elution with ethyl acetate, the column was eluted with 100 mL of 75 percent acetonitrile/25 percent water followed by 100 mL of 25 percent acetonitrile/75 percent water. The two eluates were combined, and the acetonitrile was removed by rotary evaporation. The solution from the rotary evaporator was lyophilized to yield the second sub-fraction.
Solid-state, cross-polarization magic-angle spinning (CP/MAS) 13C nuclear magnetic resonance (NMR) spectra of the samples were measured on a Chemagnetics CMX 200-megahertz (MHz) proton frequency spectrometer (Wershaw and others, 1998) equipped with a 7.5-mm Chemagnetics ceramic probe. The samples were packed in zirconia rotors and spun at 5,000 hertz (Hz). The acquisition parameters were 5 milliseconds (ms) contact time, 1-second (s) pulse delay, and 4.5-microsecond (µs) 90 degree pulse.
Infrared (IR) spectra of the fractions were measured on a Perkin-Elmer 2000 Fourier transform spectrometer. Pellets for IR analysis were prepared by triturating 5 mg of sample and 250 mg of potassium bromide with a mortar and pestle and then pressing the mixture in a die.
Amino acid analyses were performed in the Laboratory of Dr. D. A. Martens of the Agricultural Research Service, U.S. Department of Agriculture, Tucson, Arizona, as described by Martens and Loeffelmann (2003). The compositions of the carbohydrates in the fractions were determined by gas chromatographic analyses of the alditol acetates of acid hydrolyzates by V-Labs (Covington, Louisiana) using the method described by Albersheim and others (1967). A dried, weighed sample was hydrolyzed in dilute trifluoroacetic acid followed by reduction with sodium borohydride and acetylation with acetic anhydride. The resulting alditol acetates were separated by gas-liquid chromatography and identified by comparison with calibration standards. An attempt was made to estimate total carbohydrate in each sample colorimetrically by the phenol-sulfuric acid test (Dubois and others, 1956); however, reliable results could not be obtained.
Elemental analyses of isolated fractions were performed by Huffman Laboratories, Golden, Colorado, using the methods described by Huffman and Stuber (1985). The elements that were determined are listed in table 4.
The results of the water analyses are given in table 1. The only pesticide detected in the water was atrazine, and its concentration was below the long-term method detection limit (LTMDL) of 0.004 µg/L. The inorganic values, in general, also are very low; the major anions in the water are chloride, sulfate, and nitrate. In terms of counter-ion neutralizing capacity, sulfate has the highest capacity at 94 micro-equivalents/L, followed by chloride (78 micro-equivalents/L) and nitrate (19 micro-equivalents/L). The elevated sulfate and nitrate concentrations apparently are due to acid rain inputs. Lawrence and others (2001) pointed out that "Several decades of acidic deposition have led to large amounts of available nitrogen and low amounts of available calcium in the forest soils of the basin. This condition causes low pH and elevated aluminum concentrations that often exceed toxic levels for fish, and has degraded water quality throughout much of the Neversink River." The sulfate concentration (4.5 mg/L) is within the range of sulfate concentrations measured by Welsch and others (2004) in shallow groundwaters of the Neversink drainage basin. Sulfate also will deplete available soil calcium. The pH of the water in the Neversink Reservoir is 6.15.
Table 1. Results of whole-water analyses.
[ mg/L, milligrams per liter; <, less than; E, estimated; µS/cm, microSiemans per centimeter at ambient temperature; µg/L, micrograms per liter.]
| Parameter | Result | Units | |
|---|---|---|---|
| Total Organic Carbon (TOC) * | 2.9 | mg/L | |
| Alkalinity, as CaCO3 | 5 | mg/L | |
| Fluoride | <0.17 | mg/L | |
| Chloride | 2.77 | mg/L | |
| Bromide | <0.016 | mg/L | |
| N, as NH4 | E0.02** | mg/L | |
| N,as NO2 | <0.008 | mg/L | |
| N, as alkaline persulfate-nitrogen | 0.39 | mg/L | |
| N, total as NO2+NO3 | 0.267 | mg/L | |
| P, as ortho Phosphate | <0.02 | mg/L | |
| P, as alkaline persulfate-P | <0.01 | mg/L | |
| Sulfate, as SO4 | 4.5 | mg/L | |
| Residue on Evaporation (ROE), 180º C | 25 | mg/L | |
| Turbidity, (National Turbidity units) | <2 | NTU | |
| pH (laboratory measurement) | 6.15 | pH | |
| Color | 15 | Pt-Co unit | |
| Specific Conductance (eC) | 37 | µS/cm | |
| Sodium | 2.5 | mg/L | |
| Potassium | 0.28 | mg/L | |
| Magnesium | 0.604 | mg/L | |
| Calcium | 2.61 | mg/L | |
| Aluminum | 61.9 | µg/L | |
| Antimony | <0.2 | µg/L | |
| Arsenic | E0.1 | µg/L | |
| Barium | 93.8 | µg/L | |
| Beryllium | E0.05 | µg/L | |
| Boron | 17 | µg/L | |
| Cadmium | 0.04 | µg/L | |
| Chromium | E0.6 | µg/L | |
| Copper | 30.4 | µg/L | |
| Iron | 50.5 | µg/L | |
| Lead | 0.3 | µg/L | |
| Manganese | 10.4 | µg/L | |
| Mercury | <0.020 | µg/L | |
| Nickel | 0.92 | µg/L | |
| Selenium | <0.4 | µg/L | |
| Silver | <0.20 | µg/L | |
| Vanadium | 0.14 | µg/L | |
| Zinc | 14.1 | µg/L |
* Unfiltered sample analysis.
** E: Estimated. Values lie between laboratory long-term method detection limit and lowest quantitation limit statistically determined.
The yields of the fractions are given in table 2. The coarse particulate and extractable organic fractions were the least abundant fractions, and together they constituted less than 1 percent of the total mass of the fractions. These fractions were not large enough for characterization. In the most commonly used scheme for isolating NOM from natural waters, the water is acidified prior to passing it through the XAD-8 (Aiken, 1985). The column is then eluted with base; the DOM fraction obtained in this way would contain both the hydrophobic neutral fraction and the hydrophobic acid fraction. The colloidal, fine particulate, and hydrophilic acids + neutrals fractions are generally not isolated in the conventional scheme.
Table 2. Yields and organic carbon contents of the fractions
[mg, milligram; - -, no data]
| Fraction | Mass (mg) | Percent | Organic carbon | |
|---|---|---|---|---|
| mg | Percent of total | |||
| Coarse Particulate | 4.7 | 0.24 | ||
| Extractable Organic | 9.7 | 0.50 | ||
| Fine Particulate Organic | 300 | 15.4 | 48.8 | 6.5 |
| Hydrophobic Neutral | 300 | 15.4 | 164.4 | 21.9 |
| Colloid | 352 | 18.1 | 95.0 | 12.7 |
| Base | 68 | 3.5 | 22.6 | 3.0 |
| Hydrophobic Acid | 598 | 30.8 | 296.6 | 39.5 |
| Transphilic Acids + Neutral | 150 | 7.7 | 68.0 | 9.1 |
| Hydrophilic Acids + Neutral | 160 | 8.2 | 54.4 | 7.3 |
| Total | 1942.4 | 99.8 | 750.8 | |
The infrared (IR) spectra of the Neversink fractions are shown in figure 2. These spectra are similar to the infrared spectra of NOM fractions isolated from the Great Salt Lake by Leenheer and others (2004). They found that the infrared spectra of all of the NOM fractions, with the exception of the colloidal fraction, had a prominent band near 1720 cm-1 that they attributed to carboxylic acid groups. The hydrophobic neutral, hydrophilic acid + neutral, hydrophilic base, and colloidal fractions all had amide I bands near 1,660 cm-1, and the hydrophilic acid + neutral and colloidal fractions had amide II bands near 1550 cm-1. The amide II band is specific for secondary amides. Evidence of primary amides in a sample is provided by an amide I band that is more intense than the amide II band and by the presence of a broad N-H bending band between 500 to 700 cm-1. The hydrophobic neutral, hydrophilic acid + neutral, and colloidal fractions had broad bands between 3300 and 3400 cm-1 and between 1000 and 1150 cm-1 indicative of alcohols. Prominent aliphatic hydrocarbon bands at 2960, 2930, 1460, and 1380 cm-1 were present in the spectra of the hydrophobic neutral and hydrophobic acid fractions. The IR spectrum of each of the Neversink fractions will be discussed in detail in the appropriate section below.
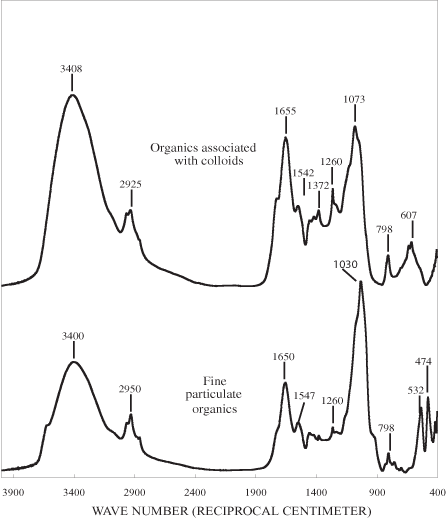
Figure 2a. Infrared spectra of particulate fractions.
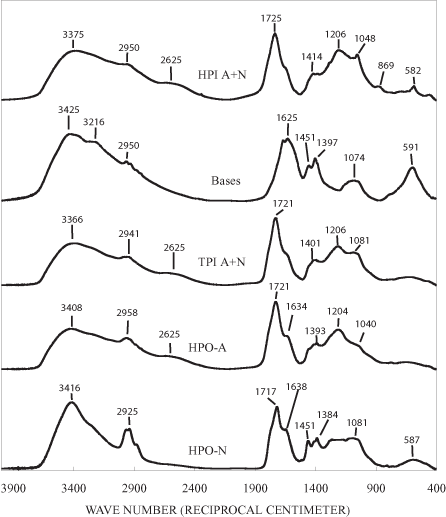
Figure 2b. Infrared spectra of dissolved fractions.
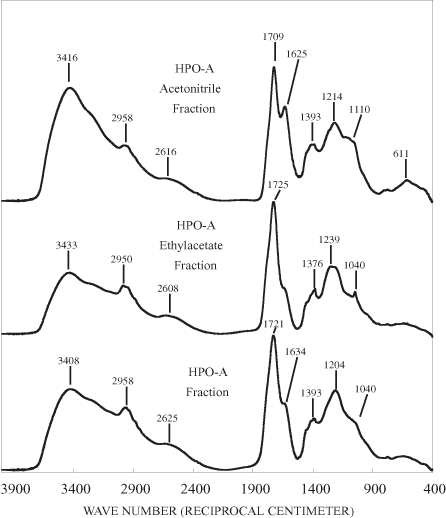
Figure 2c. Infrared spectra of HPO-A sub-fractions.
Solid-state CP/MAS 13C NMR has been used extensively in characterizing functional group distributions in NOM (Wershaw and others, 2000). The solid-state CP/MAS 13C NMR spectra of the major Neversink Reservoir NOM fractions are shown in figure 3. A generalized list of functional groups that are represented by the different regions of the NOM spectra is given in table 3. Leenheer and other (2004) and Leenheer (2004) obtained similar sets of NOM fractions from the Great Salt Lake and from Anaheim Lake; however, they did not isolate a fine-particulate organic fraction from either lake.
Figure 3a. Solid-state 13C nuclear magnetic resonance spectra of particulate fractions.

Figure 3b. Solid-state 13C nuclear magnetic resonance spectra of dissolved fractions.
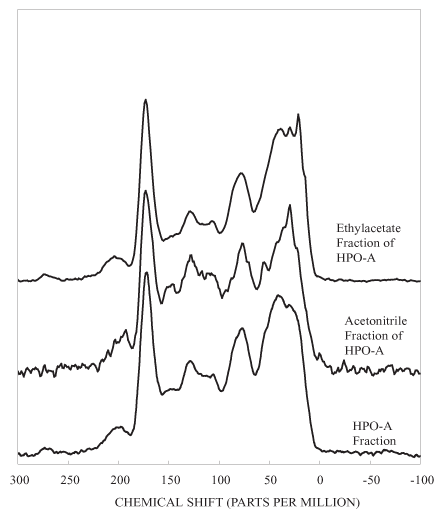
Figure 3c. Solid-state 13C nuclear magnetic resonance spectra of HPO-A sub-fractions.
[ppm, parts per million; C, carbon; O, oxygen]
| Carbon atom in functional group | Chemical shift range (ppm) |
|---|---|
| Aliphatic hydrocarbon | 0-55 |
| C adjacent to N in amines, amides, proteins | 40-55 |
| C methyl in methyl esters | 50-54 |
| C methyl in methyl ethers | 55-60 |
| C adjacent to O in alcohols, ethers | 60-90 |
| Anomeric carbons in carbohydrates, lactols | 90-110 |
| C adjacent to O in aromatic ethers, esters, phenols | 135-165 |
| Carbonyl carbons in acids, esters, amides | 160-190 |
| Carbonyl carbons in flavones, quinones | 170-200 |
| Carbonyl carbons in aliphatic or aromatic ketones | 190-220 |
Wershaw and others (2000) found that for a number of different types of NOM integration of a solid-state CP/MAS 13C NMR spectrum measured with a 5-ms contact time and a one-second pulse delay provided a semi-quantitative estimate of functional group distribution in an NOM sample. The NMR spectra of the Neversink fractions were measured using the 5-ms contact time and one-second pulse delay conditions in order to calculate functional group distributions.
The elemental analyses of the fractions are given in table 4. By combining the data on organic carbon contents of the fractions with the yield data for each fraction in table 2 the total amount of organic carbon contributed to the DOC of the Neversink water by each of the fractions can be calculated. These data are given in the third column of table 2. The total amount of carbon recovered in the entire 319 L of water in the sample was 750.8 mg. Dividing this number by 319 yields a calculated DOC value of 2.35 mg/L; the measured DOC of the sample was 2.9 mg/L, and therefore, the recovery of total DOC was 81 percent.
Table 4. Elemental analyses of fractions on an as received basis
[C, carbon; H, hydrogen; N, nitrogen; O, oxygen; FFO, fine particulate organic; HPO-N, hydrophobic neutral; HPO-A, hydrophobic acid; TPI-A+N, transphilic acid plus neutral; HPI-A+N, hydrophilic acid plus neutral]
| Fraction | C (Percent) | H (Percent) | N (Percent) | O (Percent) | C/N (Atomic) | Ash (Percent) | Moisture (Percent) |
|---|---|---|---|---|---|---|---|
| FPO | 16.64 | 3.35 | 1.95 | 18.65 | 10.0 | 60.55 | 5.53 |
| HPO-N | 54.78 | 6.41 | 1.67 | 27.33 | 38.2 | 5.72 | 2.12 |
| Colloid | 27.00 | 4.84 | 2.36 | 29.88 | 13.4 | 37.18 | 5.76 |
| Base | 33.26 | 5.36 | 9.13 | 32.56 | 4.2 | 20.79 | 5.65 |
| HPO-A | 49.61 | 5.15 | 1.05 | 42.49 | 55.0 | 1.02 | 6.06 |
| TPI-A+N | 45.25 | 5.21 | 2.22 | 44.59 | 23.7 | 1.89 | 7.31 |
| HPI-A+N | 34.01 | 4.67 | 2.89 | 50.87 | 13.7 | 5.26 | 10.93 |
The amino acid and carbohydrate analyses provide much more specific information about the chemical compositions of the fractions than any of the other characterization used in this study. The amino acid composition of each of the fractions is given in table 5. The nitrogen contribution from amino acids was calculated for each of the fractions and tabulated in table 6. In addition to the amino acid nitrogen, the nitrogen from elemental analysis also has been tabulated in the table. Comparison of the amino acid nitrogen data with the elemental analysis data indicates that all of the nitrogen in the fine-particulate fraction is derived from amino acids. Amino acid nitrogen contents were less in all of the other fractions. The colloidal fraction has the next highest contribution from amino acids, with about 67 percent of the nitrogen coming from amino acids. The other fractions have even smaller contributions from amino acids. Other nitrogen species such as ammonia, nitrate, heterocyclic nitrogen compounds, and Maillard products are probably present in some of the fractions.
Table 5. Amino acid composition of Neversink Reservoir natural organic matter.
[ Total Organic Matter, 1928 milligrams; mg/g, milligrams per gram; FPO, fine-particulate organic; HPO-A, hydrophobic acid; TPI-A+N, transphilic acid plus neutral; HPI-A+N, hydrophilic acid plus neutral; HPO-N, hydrophobic neutral. ]
| Amino Acid | FPO (mg/g) | Colloids (mg/g) | Bases (mg/g) | HPO-A (mg/g) | TPI-A+N (mg/g) | HPI-A+N (mg/g) | HPO-N (mg/g) |
|---|---|---|---|---|---|---|---|
| alanine | 10.02 | 11.00 | 0.85 | 3.85 | 3.60 | 2.86 | |
| arginine | 15.84 | 9.39 | 33.79 | 1.40 | 6.03 | 22.90 | 10.20 |
| aspartic | 13.29 | 9.51 | 12.80 | 4.73 | 5.54 | 6.66 | 3.93 |
| cysteine | 0.61 | 0.31 | 0.07 | 0.10 | |||
| diaminopimelic | 1.75 | 0.74 | 2.40 | ||||
| galactosamine | 2.13 | 4.88 | 2.75 | 0.21 | 1.18 | 1.33 | 0.48 |
| glucosamine | 4.37 | 7.56 | 3.86 | 0.89 | 2.12 | 2.10 | 1.87 |
| glutamic | 11.77 | 0.00 | 12.10 | 1.96 | 2.08 | 1.76 | 1.64 |
| glycine | 17.70 | 6.75 | 15.64 | 1.04 | 6.23 | 6.15 | 3.28 |
| histidine | 1.71 | 1.72 | 2.02 | 0.10 | 0.45 | 0.38 | 0.11 |
| hydroxyproline | 0.71 | 0.76 | 0.69 | 0.24 | 1.01 | 0.46 | 0.17 |
| isoleucine | 4.06 | 2.28 | 1.06 | 0.17 | 0.48 | 0.54 | 0.30 |
| leucine | 6.51 | 4.71 | 1.89 | 0.50 | 0.75 | 0.28 | 1.03 |
| lysine | 0.88 | 3.61 | |||||
| methionine | 0.76 | 1.29 | |||||
| muramic | 0.42 | 0.44 | |||||
| ornithine | 15.20 | 15.56 | 13.58 | 4.73 | 3.02 | 2.73 | |
| phenylalanine | 2.99 | 2.38 | 1.02 | 0.17 | 0.47 | ||
| serine/proline | 5.45 | 4.50 | 3.63 | 0.58 | 1.63 | 0.74 | 0.93 |
| threonine | 10.01 | 9.31 | 4.65 | 0.73 | 2.71 | 2.08 | 1.72 |
| tyrosine | 1.97 | 2.69 | 1.36 | 0.41 | 0.49 | 0.40 | 0.81 |
| valine | 10.74 | 7.57 | 12.54 | 9.15 | 16.40 | 16.10 | 12.10 |
| Amino Acids (mg/g) | 127.38 | 102.66 | 137.09 | 24.09 | 55.68 | 72.11 | 44.73 |
| Recovered Fraction (mg) | 300.00 | 352.00 | 68.00 | 598.00 | 150.00 | 160.00 | 300.00 |
Table 6. Contributions of amino acids to nitrogen content of fractions, as fraction totals in milligrams.
[ mg, milligram; N, nitrogen; %, percent; wt., weight; FPO, fine-particulate organic; HPO-A, hydrophobic acid; TPI-A+N, transphilic acid plus neutral; HPI-A+N, hydrophilic acid plus neutral; HPO-N, hydrophobic neutral. ]
| FPO | Colloids | Bases | HPO-A | TPI-A+N | HPI-A+N | HPO-N | |
|---|---|---|---|---|---|---|---|
| Amino Acid | N (mg) | N (mg) | N (mg) | N (mg) | N (mg) | N (mg) | N (mg) |
| alanine | 0.554 | 0.117 | 0.080 | 0.091 | 0.090 | 0.135 | |
| arginine | 1.525 | 1.061 | 0.738 | 0.269 | 0.290 | 1.176 | 0.982 |
| aspartic | 0.419 | 0.351 | 0.091 | 0.297 | 0.087 | 0.112 | 0.124 |
| cysteine | 0.025 | 0.002 | 0.005 | 0.004 | |||
| diaminopimelic | 0.083 | 0.041 | 0.026 | ||||
| galactosamine | 0.046 | 0.124 | 0.013 | 0.009 | 0.013 | 0.015 | 0.010 |
| glucosamine | 0.102 | 0.275 | 0.275 | 0.275 | 0.275 | 0.275 | 0.275 |
| glutamic | 0.275 | 0.064 | 0.091 | 0.024 | 0.022 | 0.038 | |
| glycine | 0.988 | 0.442 | 0.198 | 0.116 | 0.174 | 0.183 | 0.183 |
| histidine | 0.139 | 0.164 | 0.037 | 0.017 | 0.018 | 0.016 | 0.009 |
| hydroxyproline | 0.025 | 0.032 | 0.006 | 0.017 | 0.018 | 0.009 | 0.006 |
| isoleucine | 0.130 | 0.086 | 0.008 | 0.011 | 0.008 | 0.009 | 0.010 |
| leucine | 0.209 | 0.177 | 0.014 | 0.032 | 0.012 | 0.005 | 0.033 |
| lysine | 0.101 | 0.111 | |||||
| methionine | 0.021 | 0.043 | |||||
| muramic acid | 0.007 | 0.009 | |||||
| ornithine | 0.967 | 1.161 | 0.196 | 0.150 | 0.102 | 0.174 | |
| phenylalanine | 0.076 | 0.071 | 0.006 | 0.009 | 0.000 | 0.000 | 0.012 |
| serine/proline | 0.217 | 0.211 | 0.033 | 0.046 | 0.033 | 0.016 | 0.037 |
| threonine | 0.354 | 0.387 | 0.037 | 0.052 | 0.048 | 0.039 | 0.061 |
| tyrosine | 0.046 | 0.073 | 0.007 | 0.019 | 0.006 | 0.005 | 0.019 |
| valine | 0.387 | 0.320 | 0.102 | 0.657 | 0.295 | 0.309 | 0.436 |
| N Totals (mg) | 6.02 | 5.61 | 1.97 | 2.10 | 1.54 | 2.50 | 2.55 |
| Fraction wt. (mg) | 300 | 352 | 68 | 598 | 150 | 160 | 300 |
| Amino Acid N (%) | 2.01 | 1.59 | 2.90 | 0.35 | 1.03 | 1.56 | 0.85 |
| Elemental | |||||||
| Analysis N (%) | 1.95 | 2.36 | 9.13* | 1.05 | 2.22 | 2.89 | 1.67 |
* Includes 1.5 percent ammonia nitrogen
Carbohydrate analyses are given in table 7. There are two separate groups of fractions. Rhamnose, arabinose, and xylose are present in the fine-particulate and colloid fractions, and absent in the rest of the fractions. The meaning of these differences will be discussed in detail in the next section.
Table 7. Relative concentrations of carbohydrates in Neversink fractions
[FFO, fine particulate organic; HPO-N, hydrophobic neutral; HPO-A, hydrophobic acid; TPI-A+N, transphilic acid plus neutral; HPI-A+N, hydrophilic acid plus neutral]
| Sugar | FPO | Colloid | HPO-N | HPO-A | TPI-A+N | HPI-A+N |
|---|---|---|---|---|---|---|
| Rhamnose | 0.07 | 0.19 | 0.00 | 0.00 | 0.00 | 0.00 |
| Arabinose | 0.43 | 0.25 | 0.00 | 0.00 | 0.00 | 0.00 |
| Xylose | 0.05 | 0.19 | 0.00 | 0.00 | 000 | 0.00 |
| Mannose | 0.09 | 0.12 | 0.29 | 0.13 | 0.25 | 0.47 |
| Galactose | 0.16 | 0.17 | 0.33 | 0.27 | 0.33 | 0.24 |
| Glucose | 0.20 | 0.08 | 0.38 | 0.60 | 0.42 | 0.29 |
Chemical structural characterization of each of the fractions provides a means to infer precursor compounds and to better understand the biogeochemical processes that have altered the precursors to produce NOM. The results of NMR and IR analyses have been combined with the results of elemental, amino acid, and specific carbohydrate analyses to provide a more complete structural characterization of each fraction.
The fine-particulate organic (FPO) fraction contains the highest ash content of all of the fractions (table 4). Infrared bands at about 470 and 1,030 cm-1 indicate that silica-containing minerals are present in the fraction. The solid-state 13C NMR spectrum of the NOM in the FPO fraction consists of well-resolved bands in the aliphatic, carbohydrate, aromatic, and carbonyl regions (table 3), with the major bands being in the carbohydrate region (fig. 3a). Carbohydrate-containing polymers are abundant in terrestrial plants and in the cell walls of bacteria, fungi, and algae. In terrestrial plants, the major carbohydrates are cellulose and hemicelluloses; whereas, in bacteria peptidoglycans consisting of polysaccharide chains linked to peptides are abundant. An example of these peptidoglycans is chitin, a linear homopolysaccharide composed of (β1→4) linked N-acetyl-D-glucosamine units; it is a major component of fungi and algae cell walls. The carbohydrate bands, with the exception of the anomeric carbon band, are generally not well enough resolved in NOM samples to distinguish between these various types of carbohydrates that one would expect in NOM. The anomeric carbon bands carbohydrates are generally in the region between 90 and 110 ppm (Escuyer and others, 2001; Gilleron and others, 1997; Ramos and others, 2001; Schäffer and others, 1999). The anomeric band in the FPO fraction occurs at about 104 ppm. This is in the same region as the anomeric carbon atoms of chitin and cellulose, and the bands at 23.6, 58.1, 61.8, 72.5, 103.7, and 173.4 ppm are within 1 or 2 ppm of the bands that have been observed by Heux and others (2000) and Kono (2004) for chitin. The infrared spectrum of the fraction (fig. 2a) shows well-defined bands at 1655 reciprocal centimeters (cm-1) and 1542 cm-1; the positions of these bands are consistent with those of the amide I and amide II of chitin (Andrade and others, 2003).
The amino acid and amino sugar compositions of the fraction given in table 5, however, do not support the conclusion that chitin is the most abundant component of the NOM in the FPO fraction. Chitin is a linear polysaccharide composed of (β1→4) linked N-acetyl-D-glucosamine units, and only about 1.7 percent of the nitrogen identified in the fraction is present as glucosamine (table 6). In addition, the bands in the NMR spectra are broader than those for pure chitin; this broadening could be caused by the presence of proteins, fatty acids, and other carbohydrates and peptidoglycans along with chitin in the fine-particulate fraction. Cellulose has the same structure as chitin except that the C-2 carbon atom is attached to a hydroxyl group rather than an N-acetylamine group; that is to say, it is composed of (β1→4) linked D-glucose units. The (β1→4) linkage is also present in bacterial peptidoglycans in which alternating units of N-acetylglucosamine and N-acetylmuramic acid are linked by (β1→4) bonds (van Heijernoort, 2001). It should be noted that muramic acid, which is an indicator of bacterial peptidoglycans, is present only in the FPO and colloidal fractions.
The carbohydrate compositions of the FPO fraction and the colloid fraction differ from the other fractions in that they contain rhamnose, arabinose and xylose that are absent from the other fractions (table 7). These sugars, along with the other sugars listed in table 7, are often present in the peptidoglycan polymers that compose the outer cell walls of bacteria. Keddie and Bousfield (1980) showed that the cell walls of bacteria of the genus Corynebacterium are characterized by the presence of diaminopimelic acid (DAP), arabinose, and galactose. Minnikin and Goodfellow (1980) have listed a number of other genera of bacteria that are characterized by DAP, arabinose, and galactose in their cell walls. Schlegel (1993) pointed out that many bacteria have an outer layer of carbohydrates (capsular material) that contains glucose, aminosugars, rhamnose, 2-keto-3-galactonic acid, uronic acids of different sugars, and organic acids. The lipopolysaccharides of blue-green algae (cyanobacteria) contain galactose, glucose, mannose, xylose, arabinose, and rhamnose monomers (Cardemil and Wolk, 1976, 1979; Dunn and Wolk, 1970; Keleti and Sykora, 1982; Keleti and others, 1979; Mikheyskaya, 1977). Keleti and others (1979) pointed out that blue green algae are commonly found in drinking water supplies.
Evidence for the presence of lipids in the FPO fraction is provided by the band at 29.7 ppm in the NMR spectrum that most likely represents methylene groups in fatty acids. A shoulder in the IR spectrum of the fraction at about 1720 cm-1 probably represents the carboxylic acid groups of the fatty acids.
In addition to the carbohydrate and lipid bands, the spectrum of the FPO fraction has aromatic bands at about 128 and 150 ppm. These bands probably represent, at least in part, aromatic methylene and phenolic groups in amino acids such as phenylalanine and tyrosine.
The data outlined above have led us to conclude that the FPO fraction is composed of a mixture of chitin, peptidoglycans, and lipopolysaccharides derived from algal, bacterial, and fungal cell walls. The high ash content of the fraction is due to associated clay minerals.
The colloidal fraction is closer in composition to the FPO fraction than to any of the other fractions. As noted above, it is the only fraction other than the FPO fraction that contains rhamnose, arabinose, and xylose. Both the colloidal and FPO fractions have high ash contents, and the C/N atomic ratios are similar for the two fractions. The diagnostic silica band at 470 cm-1 is absent in the IR spectrum of the colloidal fraction (fig. 3) indicating that the high ash content is probably due to the presence of inorganic salts. The NMR spectra of the two fractions also are similar. Carbohydrate bands are the most prominent bands in both spectra; however, the aliphatic bands are considerably stronger in the FPO fraction. The chemical shift of the anomeric carbon band in the colloidal fraction is 101 ppm compared with 104 ppm in the FPO fraction. The aromatic bands are weaker in the colloidal fraction than in the FPO solids. These data indicate the colloidal fraction is composed mainly of peptidoglycans; the weaker aliphatic bands in NMR spectrum of the colloidal fraction compared with that of the FPO fraction indicate that lipids are less abundant in the colloidal fraction than in the FPO.
The NMR spectrum of HPO-N fraction is very similar to that of the HPO-N fraction isolated by Leenheer and others (2004) from the Great Salt Lake. The study by Wershaw and others (2000) showed that the use of a 5 ms contact time in CP/MAS experiments provides a reasonably quantitative spectrum. The NMR spectrum shows that the fraction consists mainly of aliphatic structural components. The band at about 74 ppm and the shoulder at about 105 ppm most likely represent carbohydrates. The strong overlapping bands in the region between 0 and 40 ppm represent aliphatic methyl, methylene, and methine groups. Distribution of functional groups estimated by integration of the NMR spectrum combined with elemental composition data were used to calculate the average number of rings and the average number of carbon atoms per ring as described by Leenheer and others (2004). These calculations yielded an average number of carbon atoms per ring of 7.7. All of these results taken together indicate that the HPO-N fraction most likely consists of a mixture of alicyclic terpenes and carbohydrates. The band at about 128 ppm in the NMR spectrum is probably indicative of olefinic and aromatic carbons in the terpenes.
The NMR spectrum of the HPOA fraction consists of relatively broad bands in all of the spectral regions (figure 3). The HPOA fraction was further fractionated on silica gel into two subfractions. The mass of subfraction 1 was 85.5 mg and that of subfraction 2 was 63.4 mg. The NMR spectra of the subfractions (fig. 3) are better resolved than the spectrum of the HPOA fraction itself. The NMR spectrum of the first, more polar fraction eluted with ethyl acetate has three well-resolved bands in the aliphatic region. These bands probably represent aliphatic and alicyclic structures of terpenes, and the band at about 128 ppm represents olefinic and aromatic carbons of terpenes (Leenheer and others, 2003). The bands at about 78 and 106 ppm indicate that the terpenoid structures are conjugated to carbohydrates. Acidic and ketonic functional groups, as indicated by the bands at 173 and 205 ppm, are attached to the terpenes. Terpenes are produced by all organisms; they are especially abundant in many types of plants, algae, and bacteria (Rodríguez-Concepción and Boronat, 2002; Kuzuyama, 2002). The terpenes in this subfraction, therefore, are probably produced both allochthonously by plants in the surrounding forests and autochthonously by microorganisms in the reservoir water.
The NMR spectrum of the acetonitrile subfraction of the HPOA is very similar to that of lignin components conjugated to carbohydrates (Wershaw and others, 2003). The band at about 55 ppm is diagnostic of methoxyl groups attached to lignin phenylpropenoid monomeric units. The aromatic phenols groups of the phenylpropenoid units are represented by the broad band between about 145 and 155 ppm and by some of the overlapping bands between about 105 and 120 ppm. Lignin-carbohydrate polymer fragments are produced during the microbial degradation of wood and bark (Wershaw, 2004). This fraction may also contain some hydrolysable and nonhydrolysable tannins. The band centered at about 195 ppm may be indicative of ketonic groups in flavonoid structures.
Leenheer and others (2004) characterized the TPI-A+N and the HPI-A+N fractions isolated from the Great Salt Lake. They measured the electrospray ionization, negative-ion mass spectra of these fractions. They concluded that the transphilic fraction is composed mainly of carboxylic acids derived from terpenoid precursors. Leenheer and others (2003) have characterized the DOM from a landfill leachate, two surface waters, and a ground water. They characterized fractions of the DOM using 13C CP/MAS NMR, tetramethylammonium hydroxide (TMAH) thermochemolysis, pyrolysis gas chromatography-mass spectrometry (GC-MS), and electrospray ionization mass spectrometry (ESI-MS), and found that terpenoid structures are major precursors of the DOM in all of these waters.
The chemical shifts of the aliphatic hydrocarbon atoms of terpenes cover a range from about 20 ppm to about 58 ppm (Breitmaier and Voelter, 1987). This range is much broader than that of most other types of aliphatic hydrocarbons. The chemical shifts of the bridgehead carbon atoms of bicyclic terpenes occur in the region between about 30 and 58 ppm. The chemical shifts of aliphatic carbon atoms attached to nitrogen in amino acids and proteins are in the region between 40 and 60 ppm, and the shifts of the carbon atoms of methyl groups of methyl ethers occur near 55 ppm. In the NMR spectrum of a DOM sample a well-defined 55-ppm band is generally associated with phenolic bands between 145 and 155 ppm; these bands are indicative of phenolic methyl ether groups in lignin units (Wershaw and others, 2003).
The NMR spectra of both the TPI-A+N and the HPI-A+N fractions have broad bands in the region between 20 and 60 ppm. The 20- to 60-ppm region of the spectrum of the TPI-A+N fraction consists of a single large band centered at about 40 ppm with four distinct shoulders at approximately 20, 25, 45, and 50 ppm. The 20- to 60-ppm region of the HPI-A+N spectrum consists of a single well-resolved band at about 20 ppm and an ill-resolved group of bands between 30 and 60 ppm. The 20-ppm band is probably due, at least in part, to terminal methyl functionalities of N-acetyl groups. Carbohydrate bands at about 73 and 103 ppm are present in the spectra of both fractions. The TPI-A+N spectrum has a well-resolved band at 128 ppm and a weak band at about 150 ppm. The band in the region near 128 ppm in the HPI-A+N spectrum is weaker and much less well resolved than that of the TPI-A+N fraction, and there is no band in the 150 ppm region. The relatively low nitrogen content of TPI-A+N fraction (table 4) means that only a small part of the intensity between 40-60 ppm can be attributed to amino acids and peptides. The higher nitrogen content of the HPI-A+N fraction indicates that more of the 40-60 ppm intensity can be attributed to amino acids and peptides in this fraction than in the TPI-A+N fraction. The relatively weak 150 ppm band in the TPI-A+N spectrum indicates that it contains some phenolic functional groups that most likely are derived from lignin or tannin structures or both. The spectra of both fractions have strong bands at about 172 ppm that are representative of carboxylic acid functional groups.
From the results outlined above, the TPI-A+N and the HPI-A+N fractions most likely represent complex mixtures of relatively low molecular weight carboxylic acids derived from terpenes, carbohydrates, and peptides. Leenheer and others (2004) have concluded that the HPI-A+N fraction they isolated from the Great Salt Lake is derived mainly from N-acetyl amino sugars; N-acetyl amino sugar components are probably also present in the Neversink HPI-A+N fraction. The Neversink TPI-A+N fraction appears to have a higher concentration of terpenoid structures and a lower concentration of amino acids and peptides than the HPI-A+N fraction.
Elemental analysis of the base fraction (table 4) shows that this fraction has the highest nitrogen content of any of the fractions (9.13 percent). The C/N atomic ratio of the base fraction is 4.2. The total amino acid nitrogen in the base fraction is 2.9 percent, and the ammonia nitrogen is 1.5 percent (table 6). About 40 percent of the amino acid nitrogen was free amino acids that were detected prior to hydrolysis; this means that the peptide nitrogen content of the fraction is about 1.7 percent (about 20 percent of the total nitrogen in the fraction). Amino acid and ammonia nitrogen account for slightly less than half (4.4 percent) of the total nitrogen in the fraction (9.13 percent). The other half of the nitrogen most likely is present as browning (Maillard) reaction products (see Wershaw, 2004, p. 19). In the browning reaction, reducing sugars react with free amino groups in amino acids and proteins to form Amadori reaction products as described by Friedman (1996). The IR spectrum of the fraction does not have a well-defined amide I or amide II band. Only peptides and other amides have the amide I and II bands; Amadori products, free amino acids, and ammonia do not have these bands. In as much as only about 20 percent of the nitrogen is present as peptides, it is not surprising that the amide bands are absent from the IR spectrum of the base fraction.
Fractionation of the DOM of the Neversink Reservoir water yielded nine fractions. Characterization by elemental, carbohydrate, and amino acid analyses, and by NMR and IR spectrometry indicated (1) that the fine-particulate organics and colloids are mainly composed of peptidoglycans, and lipopolysaccharides derived from algal, bacterial, and fungal cell walls, (2) that the HPO-N fraction most likely consists of a mixture of alicyclic terpenes and carbohydrates, (3) that the HPOA consists mainly of lignin components conjugated to carbohydrates, (4) that the TPI-A+N and the HPI-A+N fractions most likely represent complex mixtures of relatively low molecular weight carboxylic acids derived from terpenes, carbohydrates, and peptides, and (5) that the base fraction is composed of free amino acids, browning reaction products, peptide fragments, and other nitrogen-containing compounds. The coarse particulates and solvent-extractable organics were too small in size to analyze and characterize.
Aiken, G.R., 1985, Isolation and concentration techniques for aquatic humic substances, in Aiken, G.R., McKnight, D.M., Wershaw, R.L., and MacCarthy, Patrick, eds., Humic substances in soil, sediment, and water—Geochemistry, isolation, and characterization: New York, John Wiley and Sons, p.363-385.
Aiken, G.R., 1992, Chloride interference in the analysis of dissolved organic carbon by wet oxidation method: Environmental Science and Technology, v. 26, p. 2435-2439.
Albersheim, P., Nivens, D.J., English, P.D., and Karr, A., 1967, A method for the analysis of sugars in plant cell wall polysaccharides by gas-liquid chromatography: Carbohydrate Research, v. 5, p. 340-345.
American Public Health Association, 1998, Standard methods for the examination of water and wastewater (20th ed.): Washington, D.C., American Public Health Association, American Water Works Association, and Water Environment Federation, p.3-37 - 3-43.
Andrade, V.S., Neto, Benício de Barros; Fukushima, Kazutaka; and Campos-Takaki, G. M., 2003, Effect of medium components and time of cultivation on chitin production by Mucor circinelloides (Mucor javanicus IFO 4570)—A factorial study: Revista Iberoamericana de Micología, v. 20, p. 149-153.
Breitmaier, Eberhard, and Voelter, Wolfgang, 1987, Carbon-13 NMR spectroscopy—High-resolution methods and applications to organic chemistry and biochemistry: Weinheim, VCH Verlagsgesellschaft, 515 p.
Cardemil, Liliana, and Wolk, C.P., 1976, The polysaccharides from heterocyst and spore envelopes of a blue-green alga–Methylation analysis and structure of the backbones: The Journal of Biological Chemistry, v. 251, p. 2967-2975.
Cardemil, Liliana, and Wolk, C.P., 1979, The polysaccharides from heterocyst and spore envelopes of a blue-green alga—Structure of the basic repeating unit: The Journal of Biological Chemistry, v. 254, p. 736-741.
Dubois, M., Gilles, K.A., Hamilton, J.K., Rebers, P.A., and Smith, F., 1956, A colorimetric method for determination of sugars and related substances: Analytical Chemistry, v. 28, p. 350-356.
Dunn, J.H., and Wolk, C.P., 1970, Composition of the cellular envelopes of Anabaena cylindrica: Journal of Bacteriology, v. 103, p. 153-158.
Escuyer, V.E., Lety, Marie-Annick; Torrelles, J.B., Khoo, Kay-Hooi; Tang, Jyh-Bing; Rithner, C.D., Frehel, Claude; McNeil, M.R., Brennan, P.J., and Chatterjee, Delphi, 2001, The role of embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan: The Journal of Biological Chemistry, v. 276, p 48854-48862.
Faires, L.M., 1993, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of metals in water by inductively coupled plasma-mass spectrometry: U.S. Geological Survey Open-File Report 92-634, 28 p.
Fishman, M.J., ed., 1993, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of inorganic and organic constituents in water and fluvial sediments: U.S. Geological Survey Open-File Report 93-125, 217 p.
Fishman, M.J., and Friedman, L.C., 1989, Methods for determination of inorganic substances in water and fluvial sediments: U.S. Geological Survey Techniques of Water-Resources Investigations, book 5, chap. A1, 545 p.
Friedman, Mendel, 1996, Food browning and its prevention, An overview: Journal of Agricultural and Food Chemistry, v. 44, p. 631-653.
Garbarino, J.R., 1999, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of dissolved arsenic, boron, lithium, selenium, strontium, thallium, and vanadium using inductively coupled plasma-mass spectrometry: U.S. Geological Survey Open-File Report 99-093, 31 p.
Garbarino, J.R., and Damrau, D.L.,2001, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of organic plus inorganic mercury in filtered and unfiltered natural water with cold vapor-atomic fluorescence spectrometry: U.S. Geological Survey Water-Resources Investigations Report 01-4132, 16 p.
Gilleron, Martine; Himoudi, Nourrédine; Adam, Olivier; Constant, Patricia; Venisse, Anne; Rivière, Michel: and Puzo, Germain, 1997, Mycobacterium smegmatis phosphoinositols-glyceroarabinomannans—Structure and localization of alkali-labile and alkali-stable phosphoinositides: The Journal of Biological Chemistry, v. 272, p. 117-124.
Heux, L., Brugnerotto, J., Desbrières, Versali, M.-F., and Rinaudo, M., 2000, Solid state NMR for determination of degree of acetylation of chitin and chitosan: Biomacromolecules, v. 1, p. 746-751.
Huffman, E.W.D. Jr., and Stuber, H.A., 1985, Analytical methodology for elemental analysis of humic substances in Aiken, G.R., McKnight, D.M., Wershaw, R.L., and MacCarthy, Patrick, eds., Humic substances in soil, sediment, and water—Geochemistry, isolation, and characterization: New York, John Wiley and Sons, p. 433-455.
Karlsson, Torbjörn; Perrson, Per; and Skyllberg, Ulf, 2005, Extended x-ray absorption fine structure spectroscopy evidence for the complexation of cadmium by reduced sulfur groups in natural organic matter: Environmental Science and Technology, v. 39, p. 3048-3055.
Keddie, R.M., and Bousfield, I.J., 1980, Cell wall composition in the classification and identification of Coryneform bacteria in Goodfellow, M. and Board, R.G., eds., Microbiological classification and identification: London, Academic Press, p. 167-188.
Keleti, Georg, and Sykora, J.L., 1982, Production and properties of cyanobacterial endotoxins: Applied and Environmental Microbiology, v. 43, p. 104-109.
Keleti, Georg, Sykora, J.L., Lippy, E.C., and Shapiro, M.A., 1979, Composition and biological properties of lipopolysaccharides isolated from Shizothrix calcicola (Ag) Gomont (Cyanobacteria): Applied and Environmental Microbiology, v. 38, p. 471-477.
Kono, Hiroyuki, 2004, Two-dimensional magic angle spinning NMR investigation of naturally occurring chitins: Precise 1H and 13C resonance assignment of α- and β-chitin: Biopolymers, v. 75, p. 255-263.
Kuzuyama, Tomohisa, 2002, Mevalonate and nonmevalonate pathways for the biosynthesis of isoprene units: Bioscience, Biotechnology, and Biochemistry, v. 66, p. 1619-1627.
Lawrence, G.B., Burns, D.A., Baldigo, B.P., Murdoch, P.S., and Lovett, G.M., 2001, Controls on stream chemistry and fish populations in the Neversink Watershed, Catskill Mountains, New York: U.S. Geological Survey Water-Resources Investigations Report 00-4040, 16 p.
Leenheer, J.A., 2004, Comprehensive assessment of precursors, diagenesis, and reactivity to water treatment of dissolved and colloidal organic matter: Water Science and Technology: Water Supply, v. 4, p. 1-9.
Leenheer, J.A., Croué, Jean-Philippe; Benjamin, Mark; Korshin, G.V., Hwang, C.J., Bruchet, Auguste; and Aiken, G.A., 2000, Comprehensive Isolation of natural organic matter from water for spectral characterization and reactivity testing in Barrett, S.E., Krasner, S.W., Amy, G.L., eds., Natural organic matter and disinfection by-products—Characterization and control in drinking water: ACS Symposium Series 761, Washington, American Chemical Society, p. 68-83.
Leenheer, J.A., Nanny, M.A., and McIntire, Cameron, 2003, Terpenoids as major precursors of dissolved organic matter in landfill leachates, surface water, and groundwater: Environmental Science & Technology, v. 37, p. 2323-2331.
Leenheer, J.A., Noyes, T.I., Rostad, C.E., and Davidson, M.L., 2004, Characterization and origin of polar dissolved organic matter from the Great Salt Lake: Biogeochemistry, v. 69, p. 125-141.
Lindley, C.E., Stewart, J.T., and Sandstrom, M.W., 1996, Determination of low concentrations of acetochlor in water by automated solid-phase extraction and gas chromatography with mass selective detection: Journal of AOAC International, v. 79, no. 4, p. 962-966.
Madsen, J.E., Sandstrom, M.W., and Zaugg, S.D., 2003, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—A method supplement for the determination of fipronil and degradates in water by gas chromatography/mass spectrometry: U.S. Geological Survey Open-File Report 02-462, 11 p.
Martens, D.A. and Loeffelmann, K.L., 2003, Soil amino acid composition quantified by acid hydrolysis and anion chromatography–pulsed amperometry: Journal of Agricultural and Food Chemistry, v. 51, p. 6521-6529.
McLain, Betty, 1993, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of chromium in water by graphite furnace atomic absorption spectrophotometry: U.S. Geological Survey Open-File Report 93-449, 16 p.
Mikheyskaya, L.V., Ovodova, R.G., and Ovodov, Yu.S., 1977, Isolation and characterization of lipopolysaccharides from cell walls of blue-green algae of the genus Phormidium: Journal of Bacteriology, v. 130, p. 1-3.
Minnikin, D.E. and Goodfellow, M., 1980, Lipid composition in the classification and identification of acid-fast bacteria in Goodfellow, M. and Board, R.G., Microbiological classification and identification: London, Academic Press, p. 189-259.
Patton, C.J., and Kryskalla. J.R., 2003, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Evaluation of alkaline persulfate digestion as an alternative to Kjeldahl digestion for determination of total and dissolved nitrogen and phosphorus in water: U.S. Geological Survey Water-Resources Investigations Report 03-4174, 33 p.
Ramos, Ana; Boels, I.C., de Vos, W.W., and Santos, Helena, 2001, Relationship between glycolsis and exopolysaccharide biosynthesis of Lactococcus lactis: Applied and Environmental Microbiology, v. 67, p. 33-41.
Rodríguez-Concepción, Manuel and Boronat, Albert, 2002, Elucidation of the methylerythritol phosphate pathway for isopenoid biosynthesis in bacteria and plastids—A metabolic milestone achieved through genomics: Plant Physiology, v. 130, p. 1079-1089.
Schäffer, Christina; Kählig, Hanspeter; Christian, Rudolf; Schulz, Gerhard; Zayni, Sonja; and Messner, Paul, 1999, The diacetamidodideoxyuronic-acid-containing glycan chain of Bacillus stearothermophilus NRS 2004/3a represents the secondary cell-wall polymer of B. stearothermophilus strains: Microbiology v. 145, p. 1575-1583.
Schlegel, H.G., 1993, General microbiology: Cambridge, Cambridge University Press, 655 p.
Stoddard, J.D., and Murdoch, P.S., 1991, Catskill Mountains in Charles, D.F., ed., Acidic deposition and aquatic ecosystems–Regional case studies: New York, Springer-Verlag, p. 237-271.
van Heijenoort, Jean, 2001, Formation of the glycan chains in the synthesis of bacterial peptidoglycans: Glycobiology, v. 11, p, 25R-36R.
Vignati, D.A.L., Dworak, Tamara; Ferrari, Benoît; Koukal, Brahim; Loizeau, J-L, Minouflet, Marion; Camusso, M.I., Polesello, Stefano; and Dominik, Janusz, 2005, Assessment of the geochemical role of colloids and their impact on contaminant toxicity in freshwaters—An example from the Lambro–Po system (Italy): Environmental Science and Technology, v. 39, p. 489-497.
Welsch, D.L., Burns, D.A., and Murdoch, P.S., 2004, Processes affecting the response of sulfate concentrations to clearcutting in a northern hardwood forest, Catskill Mountains, New York, U.S.A.: Biogeochemistry, v. 68, p. 337-354.
Wershaw, R.L., 2004, Evaluation of conceptual models of natural organic matter (humus) from a consideration of the chemical and biochemical processes of humification: U.S. Geological Survey Scientific Investigations Report 2004-5121, 44 p.
Wershaw, R.L., and Hayes, T.M., 2001, Solubilization of anthropogenic compounds by humic substances in Clapp, C.E., Hayes, M.H.B., Senesi, N., Bloom, P.R., and Jardine, P.M., Humic substances and chemical contaminants: Madison, Wisconsin, Soil Science Society of America, p. 165-176.
Wershaw, R.L., Aiken, G.R., Leenheer, J.A., and Tregellas, J.R., 2000, Structural-group quantitation by CP/MAS 13C NMR measurements of dissolved organic matter from natural surface waters in Ghabbour, E.A. and Davies, Geoffrey, eds., Humic substances—Versatile components of plants, soil and water: Cambridge, Royal Society of Chemistry, p. 63-81.
Wershaw, R.L., Leenheer, J.A., Sperline, R.P., Song, Yuan; Noll. L.A., Melvin, R.L., and Rigatti, G.P., 1995, Mechanism of formation of humus coatings on mineral surfaces 1. Evidence for multidentate binding of organic acids from compost leachate on alumina: Colloids and Surfaces A: Physicochemical and Engineering Aspects, v, 96, p. 93-104.
Wershaw, R.L., Llaguno, E.C., Leenheer, J.A., Sperline, R.P., and Song, Yuan, 1996a, Mechanism of formation of humus coatings on mineral surfaces 2. Attenuated total reflectance spectra of hydrophobic and hydrophilic fractions of organic acids from compost leachate on alumina: Colloids and Surfaces A: Physicochemical and Engineering Aspects, v, 108, p. 199-211.
Wershaw, R.L., Llaguno, E.C., and Leenheer, J.A., 1996b, Mechanism of formation of humus coatings on mineral surfaces 3. Composition of adsorbed organic acids from compost leachate on alumina by solid-state 13C NMR: Colloids and Surfaces A: Physicochemical and Engineering Aspects, v, 108, p. 213-223.
Wershaw, R.L., Leenheer, J.A., and Kennedy, K.R., 1998, Use of 13C NMR and FTIR for elucidation of degradation pathways during natural litter decomposition and composting. III. Characterization of leachate from different types of leaves, in Davies Geoffrey and Ghabbour, E.A., eds., Humic substance—Structures, properties, and uses: Cambridge, Royal Society of Chemistry, p. 43-68.
Wershaw, R.L., Rutherford, D.W., Leenheer, J.A., Kennedy, K.R., Cox, L. G., and Koci, D.R., 2003, Biogeochemical processes that produce dissolved organic matter from wheat straw: U.S. Geological Survey Water-Resources Investigations Report 03-4213, 14 p.
Zaugg, S.D., Sandstrom, M.W., Smith, S.G., and Fehlberg, K.M., 1995, Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of pesticides in water by C-18 solid-phase extraction and capillary-column gas chromatography/mass spectrometry with selected-ion monitoring: U.S. Geological Survey Open-File Report 95-181, 60 p.
Document Accessibility: Adobe Systems Incorporated has information about PDFs and the visually impaired. This information provides tools to help make PDF files accessible. These tools convert Adobe PDF documents into HTML or ASCII text, which then can be read by a number of common screen-reading programs that synthesize text as audible speech. In addition, an accessible version of Acrobat Reader 6.0, which contains support for screen readers, is available. These tools and the accessible reader may be obtained free from Adobe at Adobe Access.
| AccessibilityFOIAPrivacyPolicies and Notices | |
 |
|