Scientific Investigations Report 2010–5154
Methods and ProceduresSample CollectionA preliminary study was done in the summer of 1998 to ascertain if stable isotope analysis might be useful in identifying what materials were the primary contributors to decomposable organic material in Tualatin River bed sediment. Bed sediment, suspended sediment, and seston (suspended particulate material, including plankton and organic detritus) samples were collected at two sites in the Tualatin River. Terrestrial materials, such as leaf litter and soil, also were collected from several sites. After the preliminary study indicated that the method might be successful in identifying the primary sources of organic matter to bed sediment, a more extensive study was designed and completed. Bed sediment, suspended sediment, and seston samples were obtained at eight sites along the Tualatin River, near the mouths of four tributaries, and from Hagg Lake during four separate sampling periods (mid‑summer-1999, late-summer-1999, winter-2000, and early-summer-2000). Not every site-material combination was collected during each sampling period. Sites were selected based on several factors, such as (a) location upstream or downstream of WWTF discharges, (b) the amount of algal growth that typically occurs in different river reaches, (c) the relative size of tributary inputs, and (d) safety considerations. Sites from RM 5.5 to 9.9 typically are influenced by large populations of algae during summer, and sites downstream of RM 38.1 are affected by discharges from major WWTFs (at RMs 38.1 and 9.3), whereas sites upstream are less affected by algal growth and WWTF discharges. Tributary sites with major contributions to the river were selected rather than sites in smaller tributaries. Finally, sites had to be safely accessible—sites that were wadeable or accessible by canoe were preferred for collecting bed sediment. In all, 92 samples were collected from July 1999 to June 2000 (fig. 2, table 1). Potential sources to the decomposable material in bed sediment also were sampled. These included various types of organic material such as leaf litter, woody material, and organic detritus. Soil samples and effluent from the two major WWTFs were collected. A sample of bryozoans (filter-feeding aquatic invertebrates) also was collected. Materials were selected that were readily found in the basin and were either known to enter the stream network or could be expected to do so. In all, 50 samples of potential source materials were collected. Bed Sediment.—The method used to collect bed sediment varied depending on the site and sampling period. For the preliminary study (late-summer-1998), a 4.2-cm diameter rigid plastic tube was used by scuba divers to obtain nine bed sediment cores from each of two sites. Core sampling was not feasible for the expanded study because some sites were accessible only by bridge. In addition, the stratigraphic information provided by cores was not needed, so a simpler method was used beginning with the mid-summer-1999 sampling period. At wadeable sites, bed sediment was collected using a stainless steel ladle to skim the top 1–2 cm of sediment from depositional areas. Areas close to the bank were avoided to reduce the contribution from nearby soil. Samples from at least three different depositional areas were obtained from each site and composited into an 8-oz glass jar. At sites accessed by canoe or bridge, the same general method was applied to a grab sample of streambed sediment collected with an Ekman dredge. Obtaining an intact streambed sample of a depositional area was difficult at some sites. Chunks of wood, concrete, asphalt, and other debris frequently caught in the jaws of the Ekman dredge and caused any fine depositional material to be washed away as the apparatus was retrieved. Some samples consisted of only compacted clay with no depositional material. For these reasons, material from only two depositional areas was composited at the most difficult sites. Bed sediment sampling in the winter proved especially difficult because high flows caused the Ekman dredge to drift and become snagged on submerged debris, jeopardizing its retrieval. Consequently, bed sediment sampling for the winter period was discontinued after only two sites were sampled. Suspended Sediment.—Suspended sediment was collected using a USGS suspended sediment sampler and the equal‑width-increment method (Edwards and Glysson, 1999). A DH-59 sampler attached to a cable was used for deep sites; a DH-81 hand sampler was used for shallow sites. A total of 4 L of water were collected at each site and stored in polyethylene bottles. Seston.—A nylon plankton tow net with an 80-μm filter mesh was used to collect seston samples. The net was slowly towed (by canoe or by hand) in the upper 1 m of the water column but below the water surface. The concentrates from each tow were composited into glass jars until approximately 1 L of concentrate was collected. Total towing time ranged from as little as 5 minutes to more than 90 minutes, depending on the amount of seston in the river or lake. Other Samples.—The method used to collect other samples depended on the sample type. In general, aggregated samples (soil, duckweed mat, leaf litter) were scooped with a stainless steel ladle, whereas discrete samples (twigs, leaves or needles, periphyton) were hand picked. All samples were placed in glass containers. Samples of wastewater effluent (50 L in stainless steel milk cans) were collected by the operators of the WWTFs. Sample ProcessingAfter collection, samples were stored at 4 ºC until processing, which generally occurred within 48 hours. Sample processing differed somewhat for bulk materials (including bed sediment, leaf litter, and soil) versus filterable material (seston, suspended sediment, and WWTF effluent). Bulk Material.—Bulk material first was examined visually for extraneous material, which was removed with forceps; for example, wood pieces were removed from bed sediment samples. Some bulk material was hand sorted into fractions, such as cones and twigs, which were separated from duckweed mat. Next, the sample was frozen and then freeze‑dried. The resulting dehydrated material was ground in a mill and portioned into three glass vials (called “sample splits”) that were sent for isotopic analysis. Filtered Material.—Suspended sediment and solids from WWTF effluent were captured on 0.45-μm glass fiber filters that had been baked at 500 ºC for 4 hours. The WWTF effluent was centrifuged before filtering to concentrate the solids. One‑half of the seston sample was filtered though a 202-μm nylon filter to remove zooplankton and any macroscopic debris. The resulting seston filtrate contained phytoplankton as well as suspended material in the 80–202 μm size range. Ten mL of this fraction was removed, preserved with formalin, and sent for algal speciation analysis. Solids from the seston filtrate and the remaining unfiltered fraction were captured separately on 0.45-μm glass fiber filters. All sample filters were frozen and then freeze-dried. Each dried filter was cut into three approximately equal wedges that were divided among three glass vials (sample splits) and sent for isotopic analysis. Summer 1998.—Sample processing differed for the preliminary study. Bed-sediment cores were frozen and then sectioned according to depth. The top 1, 2, or 3 cm of a core was used depending on the core. Bed sediment was not composited; rather, each core-section sample was treated separately. Core sections and other bulk materials were dried at 45 or 57 ºC, then ground in a mill and divided into vials. Bulk materials (other than core sections) were split into three glass vials (sample splits). Samples requiring filtration were treated the same as just described for samples collected after 1998, except that the filters were dried at 45 or 57 ºC. Filters were cut into three sections which were divided among vials (sample splits). Analytical MethodsStable Isotope and Elemental AnalysisSample splits were analyzed for carbon and nitrogen isotopic and elemental composition by Dr. Carol Kendall’s laboratory at the USGS facility in Menlo Park, California. Their method will be summarized here; for a detailed description, see Kendall and others (2001). Bulk sample splits were subsampled as received. Sample splits on filters were scraped into a dish and then ground before subsampling. Approximately 2 mg subsamples were used for highly organic samples (seston and plant material) and 18–25 mg subsamples were used for samples that contained less organic matter (bed and suspended sediment and WWTF effluent). Following their standard protocols, the subsamples were vapor acidified to remove carbonate and then analyzed using a Carlo Erba® elemental analyzer attached to a Micromass Optima mass spectrometer. Carbon and nitrogen isotopic compositions generally are expressed as a δ value in units of per mil (parts per thousand). A δ value is calculated relative to standards, carbon in PeeDee belemnite (PDB) and nitrogen in air (see equation 1). A substance with a higher δ value contains relatively more of the heavier isotope (13C or 15N) and relatively less of the lighter isotope (12C or 14N) than a substance with a lower δ value. The changes in percent composition are very small—a 1 per mil change corresponds to a 0.0011 and 0.0004 percent change in isotope composition for δ13C and δ15N, respectively. Carbon and nitrogen content was measured for all samples. For samples collected on filters, however, an unknown amount of glass fiber was included when the filter was scraped, thus making the percent carbon and percent nitrogen values individually meaningless. The C/N ratio, however, is valid and that is the value reported here as an atomic ratio. Quality Assurance.—Dr. Kendall’s laboratory routinely analyzes approximately 10 percent of samples in duplicate and analyzes additional splits if results are not within their expected reproducibility. About 15 percent of the analyses for this study were laboratory splits (“lab splits”). As described, all Tualatin River basin samples (except the 1998 core sections) were submitted to the laboratory as triplicate sample splits. The variance of lab splits was not significantly different from the variance of sample splits (α=0.05, F-test). If the sample material had not been homogeneous before being divided into triplicate splits, the pooled variance of the sample splits would have exceeded that of the lab splits. The fact that the variances are not different is an indication that all splits (sample and lab) were equivalent. Therefore, lab split and sample split results were treated identically. Some of the Tualatin River basin sample results, especially for bed sediment and suspended sediment samples, had poorer than expected reproducibility for δ15N. Laboratory results indicated that these samples typically contained little nitrogen, less than 1 micromole (14 μg). Nitrogen content was low enough in some cases that it was near the detection limit of the analytical instrument, causing the measurement to have only one significant digit. This resulted in higher variability among C/N ratios for the sample. After the first sets of samples from the Tualatin River basin had been analyzed, it was evident that acidification was affecting δ15N, but not δ13C, which was exactly the opposite of the desired effect. Samples typically are acidified to prevent carbonate from affecting the δ13C of the organic carbon in the sample, and the acidification should not alter the δ15N. Evidently, samples from the Tualatin River basin contained little carbonate and particularly labile nitrogen (C. Kendall, written commun., February 25, 1999). For a number of subsequent samples, both acidified and unacidified subsamples were analyzed. These results confirmed the previous observations. Only unacidified subsamples were analyzed for the last sampling period (early-summer-2000). The inconsistency in acidification procedures resulted in data that were not necessarily comparable. For about 50 percent of the samples, all subsamples were acidified before analyses. For about 30 percent of the samples, no subsamples were acidified. The remaining samples had some, but not all subsamples, acidified before analysis. Making these data compatible with each other required devising a method to remove the artifact associated with acidification; this method is described later in this report (see section, “Acidification Correction”).
Other AnalysesAlgal speciation and enumeration was performed by Aquatic Analysts (Friday Harbor, Washington) on formalin‑preserved samples by microscopic examination and counting at least 500 colonies. Chlorophyll-a concentrations in river water were measured by the Clean Water Services Water-Quality Laboratory (Hillsboro, Oregon) as part of a routine monitoring program. Samples were collected weekly; interpolation was used for dates that did not coincide with a routine sampling date. Clean Water Services participates in a quality assurance program for chlorophyll-a analysis with the USGS Oregon Water Science Center. Statistical MethodsSeveral statistical methods were used in this study. Parametric methods (ordinary least squares, linear regression, mean, standard deviation, and Student’s t-test) were used to evaluate results of sample splits, which can be expected to be normally distributed. For comparisons among sample results, non-parametric methods were used, including analysis of variance on ranked data, Mann-Whitney U test, and Spearman rank-order regression (Miller and Miller, 1988; SAS Institute Inc., 1989; Helsel and Hirsch, 1992). A criterion of α=0.05 was used for statistical significance unless otherwise noted. Principal components analysis was performed on normalized data (SAS Institute Inc., 1989). Data Aggregation and ManipulationTriplicate sample splits were submitted for almost all samples, with the intent to calculate a mean value and confidence limits for each sample. Two inconsistencies related to changes in methods of sample collection and analytical procedures resulted in data that were not necessarily compatible among splits and could complicate comparisons among samples. Specifically,
The following protocol was devised to produce a final dataset that was as internally consistent as possible.
Acidification CorrectionLaboratory personnel judged that acidified splits produced the most reliable results for δ13C and percent carbon, and that unacidified splits were better for δ15N and percent nitrogen. Because acidified and unacidified splits were not analyzed for every sample, it was necessary to devise a correction factor for acidification. Data from the 32 samples having both acidified and unacidified results were examined. The mean value was used when multiple lab or sample splits had been analyzed, otherwise discrete values were used. Analysis of acidified versus unacidified data regressions (fig. 3) showed that the δ13C regression line was not significantly different from the y=x line, but the δ15N and C/N regressions were significantly different from the y=x line (α=0.05). These results confirmed previous observations: acidification did not significantly affect δ13C, but systematically decreased δ15N. Acidification increased C/N slightly, which is consistent with the hypothesis that some of the nitrogen was labile. Data shown in figure 3 represent samples of bed sediment, suspended sediment, seston, plant material, and WWTF effluent solids. When the effect of acidification on δ15N was first observed, it was thought to be limited to the sediment samples. Treating sediment samples separately from the others, however, showed similar regression statistics between acidified and unacidified results; therefore, all types of samples were included. Regression equations were applied to all acidified results for δ15N and C/N, correcting them to the equivalent of unacidified results (table 2). No correction was applied to δ13C results because the regression line was not significantly different from y=x. Standard Deviation AdjustmentAs previously described, the mean and standard deviation of all splits was calculated for each sample (a weighted method was used for bed-sediment core sections) after δ15N and C/N values were corrected for the effects of acidification. For δ15N and C/N results in the case where all splits were acidified, the calculated standard deviation underestimates the potential variability because it does not include the variability associated with the acidification correction regression. To remedy this situation, additional variance was added to the variance for the acidified samples:
Confidence intervals of the sample means provide guidance for interpreting differences; these intervals were calculated using a two-tailed Student’s t-test at the 95-percent level. The mean 95-percent confidence limits are shown in table 3. |
First posted August 17, 2010 For additional information contact: Part or all of this report is presented in Portable Document Format (PDF); the latest version of Adobe Reader or similar software is required to view it. Download the latest version of Adobe Reader, free of charge. |

![]() U.S. Department of the Interior |
U.S. Geological Survey
U.S. Department of the Interior |
U.S. Geological Survey
URL: http://pubsdata.usgs.gov/pubs/sir/2010/5154/section3.html
Page Contact Information: GS Pubs Web Contact
Page Last Modified: Thursday, 10-Jan-2013 19:15:16 EST

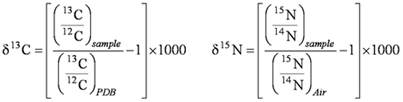 (1)
(1)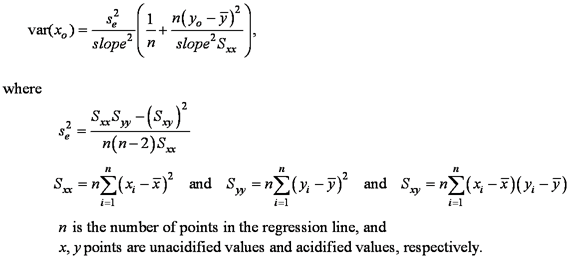 (2)
(2)