FIRE and MUD Contents
Preeruption Vapor in Magma of the Climactic Mount Pinatubo Eruption: Source of the Giant Stratospheric Sulfur Dioxide Cloud
By Terrence M. Gerlach,1 Henry R. Westrich,2 and Robert B. Symonds1
1U.S. Geological Survey, Cascades Volcano Observatory, 5400 MacArthur Blvd., Vancouver WA 98661.
2Sandia National Laboratory, Geochemistry Dept. 6118, Albuquerque NM 87185.
ABSTRACT
The climactic June 15, 1991, eruption of Mount Pinatubo injected a minimum of 17 Mt (megatons) of SO2 into the stratosphere -- the largest stratospheric SO2 cloud ever observed. This study is an investigation of the immediate source of the sulfur for the giant SO2 cloud. Approximately 100 electron microprobe analyses show no significant differences, at the 95 percent confidence level, in S or Cl contents between glass inclusions and matrix glasses of the erupted dacite. These results indicate that there was no significant degassing of S or Cl from melt during ascent and eruption. Furthermore, the 17-Mt SO2 cloud contained over an order of magnitude more sulfur than could have been dissolved in the quantity of erupted silicate melt at the preeruption conditions. A major source of "excess sulfur" is therefore required to account for the SO2 cloud. Degassing of melt in non-erupted dacite as a source of the excess sulfur implies volumes of non-erupted dacite larger than the estimated volume of the magma reservoir beneath the Mount Pinatubo region. Direct degassing of excess sulfur from a basalt source seems unlikely, since the June 15 eruption products lack evidence of mixed or commingled contemporaneous basalt. Anhydrite decomposition rates at atmospheric pressure and expected eruption temperatures are extremely slow and grossly incapable of generating 17 Mt of SO2 by anhydrite breakdown in the eruption cloud. Anhydrite breakdown during ascent decompression is too slow to keep pace with conduit travel times, which were considerably less than 8 minutes. Flash vaporization of sulfate-rich Pinatubo hydrothermal fluids during the eruption could have caused sulfate mineral deposition but virtually no SO2 production.
It is proposed that the dacite erupted on June 15 was vapor-saturated at depth prior to eruption, and that an accumulated vapor phase in the dacite provided the immediate source of excess sulfur for the 17-Mt SO2 cloud. Investigations based on exploration drilling for geothermal energy suggest that magmatic volatiles were discharged into the Pinatubo hydrothermal system from the vapor-saturated dacite prior to the 1991 eruption. Experimental studies, geobarometer results, and the H2O and CO2 contents of glass inclusions indicate that the Pinatubo dacite was saturated with water-rich vapor before ascent and eruption. Models for the composition of the preeruption vapor suggest that it contained a minimum of approximately 96 Mt H2O, 42 Mt CO2, and 3 Mt Cl, in addition to 17 Mt of SO2. The mole fraction composition of the vapor was XH2O = 0.80 - 0.83, XSO2 = 0.01 - 0.04, XCO2 = 0.15, and X
Cl = 0.01, indicating that the vapor was not excessively SO2-rich. The volume and density of the vapor at depth prior to eruption were >=0.25 km3 and about 0.6 g/cm3, respectively. Vapor comprised at least 5 volume percent of the preeruption dacite at depth; the bulk density of the preeruption dacite was less than 2.3x1012
kg/km3. Solubility modeling indicates that the total amount of volatiles contained in the preeruption vapor and melt of the erupted dacite could not have been dissolved initially in completely molten dacite at the magma reservoir pressures, suggesting some process of preeruption vapor accumulation at depth.
The climactic eruption released the accumulated vapor in the estimated 5 km3 of erupted dacite. About 6.25 wt percent of dissolved water was also degassed from melt during ascent and eruption; scaling to the volume of erupted dacite implies an additional release of 395 Mt of H2O. Additional yields of SO2, CO2, and Cl from degassing of melt were minor to insignificant during ascent and eruption. Thus, the minimum volatile emissions for the climactic eruption -- from preeruption vapor phase and degassing of melt -- were 17 Mt SO2, 42 Mt CO2, 3 Mt Cl, and 491 Mt H2O.
This study underscores the need for both petrologic measurements and emission measurements to constrain the quantity of dissolved volatiles and preeruption vapor in magma at depth. If explosive volcanism commonly involves magmas with substantial accumulated vapor, the volatile contents of glass inclusions alone are not a sufficient basis for inferring the total preeruptive volatile contents of magma and for predicting volatile emissions. Consequently, conventional petrologic estimates of SO2 emissions during explosive eruptions of the past may be far too low and significantly underestimate their impacts on climate and the chemistry of the atmosphere.
Note to readers: Figures open in separate windows. To return to the text, close the figure's window or bring the text window to the front.
INTRODUCTION
Scientists have used remote sensing to measure volcanic SO2 emissions by correlation spectrometry (COSPEC) since 1972 (Stoiber and Jepsen, 1973) and by Total Ozone Mapping Spectrometry (TOMS) from the Nimbus-7 satellite since 1978 (Bluth and others, 1993). Petrologic analysis of eruption products is used to estimate SO2 emissions and to assess atmospheric and climatic impacts of eruptions occurring before COSPEC and TOMS data were available. The conventional petrologic method assumes that the volatile contents of glass inclusions trapped in crystals are representative of the preeruption melt, and that the volatile contents of coexisting degassed matrix glass represent the melt after eruption. The difference in the volatile contents of the glass inclusions and matrix glasses is taken as a measure of volatile degassing. Scaling up volatile degassing for the mass of erupted melt yields estimates of volatile emissions during eruption (Johnston, 1980; Devine and others, 1984; Sigurdsson and others, 1985; Palais and Sigurdsson, 1989; Sigurdsson, 1990).
The petrologic method may tend to underestimate volatile emissions, since assumptions inherent in the method, if not fully satisfied, lead to low results (Devine and others, 1984; Palais and Sigurdsson, 1989). Comparisons of COSPEC and TOMS results with petrologic estimates for explosive eruptions from subduction zone volcanoes suggest that petrologic estimates of sulfur emissions are low by more than an order of magnitude (Luhr and others, 1984; Sigurdsson, 1990; Sigurdsson and others, 1990; Williams and others, 1990; Andres and others, 1991; Gerlach and others, 1994), although comparisons have been limited to explosive eruptions with a Volcano Explosivity Index (VEI) <=5. An important issue is whether or not the level of agreement between petrologic and remote sensing results improves for larger eruptions (Sigurdsson, 1990). The June 15, 1991, climactic eruption of Mount Pinatubo permits a comparison of petrologic emission estimates for SO2 with remote sensing determinations for a larger eruption (VEI=6) that had a clear impact on climate and stratospheric ozone (Dutton and Christy, 1992; Hofman and others, 1992; Prather, 1992; Gleason and others, 1993; Halpert and others, 1993; Solomon and others, 1993; McCormick and others, 1995).
This study is an investigation of the sources of sulfur for the giant SO2 cloud injected into the stratosphere on June 15, 1991, during the climactic eruption of Mount Pinatubo. It is confined to immediate sources -- that is, sources of sulfur potentially available at the moment the eruption began. We show that conventional petrologic estimates fail unconditionally in predicting the quantity of SO2 released during the climactic eruption. Contrary to expectation, the preeruption melt was not a significant source of sulfur for the giant cloud. We analyze several alternative sources of sulfur and conclude that the SO2 of the stratospheric cloud was the product of vapor saturation and accumulation in the Pinatubo magma at depth prior to eruption.
SO2 EMISSION ESTIMATES BY REMOTE SENSING METHODS
Table 1 summarizes several attributes of the climactic June 15 eruption. The climactic event got underway at 1342 and lasted ~9 hours until 2230 (Wolfe and others, this volume; Hoblitt and others, this volume). It erupted 4-5 km3 dense-rock equivalent (DRE) (W.E. Scott and others, this volume) of dacite magma (Pallister and others, this volume) and produced the largest SO2 cloud ever observed in the stratosphere. Three determinations of the SO2 content of the stratospheric cloud are now available from remote sensing data. The TOMS determination is 20+-6 Mt (20 megatons= 20x109 kg) (Bluth and others, 1992). This result may include ~0.5 Mt of SO2 from explosions preceding the climactic event (Bluth and others, 1992), an amount well within the estimated error. The Microwave Limb Sounder (MLS) experiment on the Upper Atmosphere Research Satellite (UARS), launched into orbit by the Space Shuttle on September 12, 1991, measured ~0.9 Mt of residual SO2 100 days after the climactic eruption (Read and others, 1993). A best-fit decay line to this and subsequent MLS/UARS measurements recording the conversion of SO2 to sulfuric acid gives an extrapolated initial SO2 injection of 17 Mt (Read and others, 1993). The total error in this extrapolated value is not known but is assumed to be greater than +-1 Mt (W. G. Read, oral commun., January 28, 1994). The third remote sensing determination comes from spectral scan data of the SBUV/2 instrument on the NOAA-11 satellite. SBUV/2 data give an initial SO2 injection of 13.5+-1.5 Mt (McPeters, 1993). The unweighted mean of these results is 17 Mt with an estimated error in the mean (s/n1/2) of +-2 Mt.
In this report, we take the 17 Mt mean remote sensing value for the SO2 content of the cloud injected into the stratosphere as a minimum estimate of the total SO2 emission during the climactic eruption. We assume that early removal of SO2 may have occurred by scavenging on ash. It is estimated, for example, that nearly 30% of the vaporous sulfur in the 1982 eruptions of El Chichón Volcano (Varekamp and others, 1984) and over 30% of the sulfur in the 1974 eruptions of Fuego Volcano (Rose, 1977) were scavenged by ash within the plume.
Table 1. Attributes of the climactic June 15, 1991, eruption, Mount Pinatubo.
[MPa, megapascal; Mt, megaton, 109kg; DRE, dense rock equivalent; NNO, O2 fugacity of the nickel-nickel oxide buffer]
|
Parameter
|
Value
|
Basis
|
|
Eruption duration (te)
|
9 hours
|
Seismic and barograph records1
|
|
DRE volume erupted magma (V)
|
5 km3
|
Studies of eruption deposits and topographic changes2
|
|
Total SO2 emission (ESO2)
|
17+-2 Mt (minimum)
|
TOMS, MLS, and SBUV/2 remote sensing3
|
|
Magma O2 fugacity (fO2)
|
10-12 MPa (NNO+3.2)
|
Fe-Ti oxides and experimental calibration4
|
|
Magma temperature (T)
|
780+-10°C
|
Fe-Ti oxides and experimental calibration4
|
|
Preeruption pressure (P)
|
220+-50 MPa
|
Al-content hornblende and experimental phase equilibria4
|
|
DRE melt volume fraction (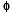 m) m)
|
0.55
|
Groundmass volume fraction of white pumice by point-count, vesicle-free basis5
|
|
Melt density (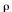 m) m)
|
2.3x1012 kg/km3
|
Rhyolite glass with 6 weight percent water6
|
|
Dacite density ()
|
2.4x1012 kg/km3
|
Estimated for crystal-glass of dacite, vesicle-free basis2
|
|
Magma reservoir depth (d)
|
6-11 km
|
Inversion of seismicity data7
|
SO2, Cl, AND H2O EMISSION ESTIMATES BY THE PETROLOGIC METHOD
SAMPLES AND PROCEDURES
The correlation of the giant stratospheric SO2 cloud with the dacite magma erupted on June 15 implicates the dacite as the likely source of the SO2. We therefore focus on the June 15 dacite in our determination of petrologic emission estimates for SO2. The climactic June 15 eruption produced two dacite lithologies (Pallister and others, 1992; this volume). The dominant lithology is a white, phenocryst-rich (45 volume percent crystals) hornblende dacite pumice with 64-65 wt percent SiO2 (anhydrous whole-rock basis). The subordinate lithology is a grayish-tan, phenocryst-poor dacite pumice with a similar whole-rock composition but a texture characterized by abundant micrometer-sized broken crystals suggesting mechanical fragmentation and shattering of former phenocrysts. The phenocryst-rich dacite comprises ~85 percent of the June 15 pyroclastic flow deposits. Inclusions of the phenocryst-rich pumice are common in deposits of the phenocryst-poor pumice, and some deposits contain the two types intermingled as banded pumice blocks. In addition to similar whole-rock compositions, the two dacite pumice types have similar phenocryst assemblages, although the phenocryst content is much lower in the fragmented phenocryst-poor pumice. Hornblende, with cummingtonite rims on some crystal faces, and plagioclase are the main phenocrysts in both dacites, and both types also contain anhydrite, magnetite, quartz, and apatite phenocrysts; sulfides are rare (Imai and others, 1993; Fournelle and others, this volume; Hattori, this volume). See Pallister and others (1992; this volume) for modal petrographic data.
Our samples were collected from rapidly cooled tops of pyroclastic flow deposits within six days of the climactic June 15 eruption. They include both the phenocryst-rich and phenocryst-poor dacite pumice of pyroclastic flow deposits located northeast of Mount Pinatubo in the Sacobia River valley and to the northwest in the Bucao River valley. The glass inclusion analyses reported here come from both the phenocryst-rich and phenocryst-poor dacite pumice. The matrix glass analyses are restricted to the phenocryst-rich dacite, since it contains clear groundmass glass suitable for electron microprobe analysis. Microscopic crystal fragments make electron microprobe analysis of matrix glass difficult in the phenocryst-poor dacite (Pallister and others, this volume).
Light crushing and sieving followed by suspension in heavy liquids allowed separation and concentration of phenocrysts from the glassy matrix of the pumice samples. Phenocrysts were mounted in epoxy and their glass inclusions were exposed by grinding and polishing. To identify the glass inclusions, we examined the polished phenocryst surfaces with reflected light and backscattered electron microscopy. Glass inclusion-bearing phenocrysts include hornblende, magnetite, plagioclase, and quartz. Most glass inclusions are clear to brown in color and vary in size from <80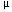 m in hornblende, magnetite, and plagioclase to 300 m in quartz.
m in hornblende, magnetite, and plagioclase to 300 m in quartz.
Major element analyses of glass inclusions and matrix glasses were obtained on a JEOL JXA-8600 electron microprobe at an accelerating voltage of 15 kV, a beam current of 10 nA, a beam diameter of 10 m, and standard data reduction techniques (Bence and Albee, 1968). A higher beam current (20 nA) and longer counting times (80-120 s) improved detection limits for chlorine (Cl, 40 ppm) in matrix glass and glass inclusions and for sulfur (S, 55 ppm) in glass inclusions. Analytical precision for Cl and S was better than +-10 percent. Wavelength measurements of S-K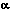 X-rays gave the proportions of sulfur present as sulfate and sulfide (Carroll and Rutherford, 1988). A Cameca IMS 3f ion microprobe with a 1 nA mass-analyzed primary beam of 16O- ions provided analyses of selected glasses for H2O and several trace elements. Secondary ions from a 20 m spot were collected using a voltage offset (Hervig and Williams, 1988). The ion probe detection limit for water in glasses is 0.15 wt percent.
X-rays gave the proportions of sulfur present as sulfate and sulfide (Carroll and Rutherford, 1988). A Cameca IMS 3f ion microprobe with a 1 nA mass-analyzed primary beam of 16O- ions provided analyses of selected glasses for H2O and several trace elements. Secondary ions from a 20 m spot were collected using a voltage offset (Hervig and Williams, 1988). The ion probe detection limit for water in glasses is 0.15 wt percent.
GLASS ANALYSES
Table 2 presents mean compositions based on analyses of 86 glasses, including matrix glasses and glass inclusions in hornblende, magnetite, plagioclase, and quartz phenocrysts. The glass compositions are similar regardless of the sample site, host phenocryst, or dacite lithology they represent. When normalized to anhydrous compositions, all glasses are high silica rhyolite compositions. Their similarity, even at trace element levels (Westrich and Gerlach, 1992), indicates a comagmatic origin for the glass inclusions and the matrix glasses, and suggests that crystallization was not significant after glass inclusion entrapment. The variability of SiO2 in the glass inclusion data (table 2) reflects differences in water contents, as discussed below.
Most glasses have sulfur contents too low for precise measurement of the wavelength of S-K X-rays. A few glasses with higher than average sulfur, however, indicate sulfur is dissolved as sulfate (Carroll and Rutherford, 1988). This result is consistent with oxygen fugacity estimates (Rutherford and Devine, this volume) for the preeruption June 15 dacites of 3.2 log units above the nickel-nickel oxide buffer (NNO+3.2, table 1) (Huebner and Sato, 1970). We therefore report the sulfur data (table 2) as wt percent SO3, but also show the data converted to ppm (wt) S, which are the units commonly used in this report. Figure 1 summarizes the sulfur data from table 2 and includes 15 additional sulfur analyses. Figure 2 summarizes the Cl data from table 2 and includes 12 additional Cl analyses. Table 2 presents the mean and maximum H2O concentrations based on ion probe analysis.
Table 2. Analyses of glass inclusions and matrix glasses from the June 15, 1991, dacite pumice, Mount Pinatubo.
[Values in weight percent unless otherwise indicated. Values are means; parentheses contain standard deviations. GI, glass inclusion from hosts in pheno-cryst-rich and phenocryst-poor pumice; MG, matrix glass from phenocryst-rich pumice; Hnb, hornblende; Mgt, magnetite; Plag, plagioclase; Qtz, quartz. FeO analyzed as ferrous iron. Na2O data are not adjusted for sodium-loss during electron beam excitation. Sulfur analyzed as SO3; ppm S calculated by multiplying percent SO3 by 4,000. N is number of electron microprobe analyses. NH2O is number of ion probe analyses for H2O. H2O (max) is the maximum water content determined in the NH2O ion probe analyses.]
|
Oxide
|
Hnb GI
|
Mgt GI
|
Plag GI
|
Qtz GI
|
MG
|
|
SiO2
|
77.06 (0.56) |
76.59 (1.42) |
75.15 (1.11) |
75.61 (1.28) |
78.53 (0.94) |
|
TiO2
|
0.14 (0.03) |
0.29 (0.03) |
0.11 (0.03) |
0.10 (0.03) |
0.13 (0.03) |
|
Al2O3
|
12.88 (0.27) |
12.44 (0.21) |
12.44 (0.56) |
11.83 (0.37) |
12.86 (0.12) |
|
FeO
|
1.11 (0.11) |
1.27 (0.12) |
0.71 (0.13) |
0.61 (0.09) |
1.01 (0.15) |
|
MnO
|
0.05 (0.02) |
0.03 (0.02) |
0.03 (0.02) |
0.04 (0.02) |
0.04 (0.02) |
|
MgO
|
0.19 (0.03) |
0.19 (0.01) |
0.17 (0.05) |
0.15 (0.03) |
0.21 (0.01) |
|
CaO
|
1.27 (0.09) |
1.17 (0.03) |
1.04 (0.11) |
1.08 (0.06) |
1.21 (0.04) |
|
K2O
|
2.94 (0.10) |
2.79 (0.12) |
2.96 (0.09) |
2.82 (0.08) |
2.99 (0.07) |
|
Na2O
|
3.17 (0.24) |
2.84 (0.22) |
2.45 (0.24) |
2.70 (0.41) |
3.18 (0.21) |
|
P2O5
|
0.04 (0.02) |
0.03 (0.02) |
0.03 (0.01) |
0.02 (0.01) |
0.03 (0.02) |
|
SO2
|
0.017 (0.010) |
0.018 (0.008) |
0.022 (0.017) |
0.022 (0.014) |
0.015 (0.008) |
|
Cl
|
0.109 (0.010) |
0.093 (0.009) |
0.106 (0.025) |
0.088 (0.014) |
0.112 (0.012) |
|
H2O
|
1.34 (0.88) |
3.51 (1.54) |
3.09 (0.87) |
4.37 (1.03) |
0.31 (0.15) |
|
Total
|
100.32 |
101.26 |
98.31 |
99.44 |
100.63 |
|
N
|
25 |
12 |
10 |
18 |
21 |
|
NH2O
|
8 |
5 |
4 |
10 |
3 |
|
S (ppm)
|
68 (40) |
72 (32) |
88 (68) |
88 (56) |
60 (32) |
|
H2O (max)
|
3.30 |
4.55 |
3.52 |
6.56 |
0.43 |
Figure 1. Sulfur concentrations for the June 15 pumice matrix glasses (MG) and glass inclusions in host phenocrysts of hornblende (Hnb), magnetite (Mgt), plagioclase (Plag), and quartz (Qtz). Dotted line at 55 ppm S is the microprobe detection limit (MDL). Solid squares are mean values; error bars are +-2 standard errors (s/n1/2); and n is the number of samples. The mean values of glass inclusions and matrix glasses are not significantly different at the 95-percent confidence level, thus implying zero petrologic emission estimates for SO2.
Figure 2. Chlorine concentrations for the June 15 pumice matrix glasses (MG) and glass inclusions in host phenocrysts of hornblende (Hnb), magnetite (Mgt), plagioclase (Plag), and quartz (Qtz). Solid squares are mean values; error bars are +-1 standard deviation; and n is the number of samples. The mean values of glass inclusions and matrix glasses are not significantly different at the 95 percent confidence level, thus implying zero petrologic emission estimates for chlorine.
PETROLOGIC EMISSION ESTIMATE FOR SO2
The glass inclusion and matrix glass data for sulfur together with melt volume fractions, melt density, and volumes of erupted magma constrain petrologic SO2 emission estimates for comparison with the 17-Mt remote sensing result. This comparison assumes that sulfur was degassed almost entirely as SO2 during the eruption, or if not, that the degassed sulfur species converted rapidly to SO2, the sulfur gas species detected by TOMS and MLS. The predominance of SO2 in degassed sulfur seems likely, since the June 15 dacite had an exceptionally high oxygen fugacity (NNO+3.2, table 1), and SK X-ray data for the glasses indicate a predominance of sulfate over sulfide in the melt. Furthermore, thermodynamic calculations for the vapor phase reaction
H2S(g) + 3/2O2(g) = H2O(g) + SO2(g) (1)
indicate an equilibrium SO2/H2S fugacity ratio >55 during degassing from preeruption conditions (table 1) to atmospheric pressure (fig. 3). Similar calculations for the reaction
Figure 3. fSO2/fH2S versus total pressure during degassing of the June 15 dacite from a preeruption pressure of 220 MPa to atmospheric pressure at the dacite preeruption temperature (780°C) (table 1). The results indicate that fSO2/fH2S was always >55 during degassing. The fugacity ratio at equilibrium during degassing is given by
log(fSO2/fH2S) = logK1 + 1.5 log(fO2) - log(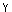 H2O) - log(XH2O) - logP
H2O) - log(XH2O) - logP
where K1=1021.63 is the equilibrium constant (Chase and others, 1985) for reaction (1) at 780°C, fO2 is the oxygen fugacity of the preeruption dacite (10-12.1 MPa, table 1), H2O is the fugacity coefficient for H2O in the gas, XH2O is the mole fraction of H2O in the gas, and P is the total pressure during degassing. Note that fSO2/fH2S increases as XH2O decreases. H2O estimated from fugacity coefficients for pure H2O (Burnham and others, 1969).
SO2(g) + 1/2O2(g) = SO3(g) (2)
indicate an equilibrium SO2/SO3 fugacity ratio of 3.5x105
during degassing. These calculations support the assumption that sulfur was degassed predominantly as SO2 during the June 15 eruption.
The equation for calculating petrologic emission estimates for SO2 (ESO2) is as follows:
ESO2 = 2(10-15) 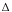 Sm
mmV (3)
Sm
mmV (3)
where Sm is the melt S-loss during eruption taken as the difference in the S contents of glass inclusions and matrix glasses in ppm, m is the bubble-free melt density in kg/km3, m is the bubble-free melt volume fraction, and V is the DRE volume of erupted magma in km3. The constant 10-15 gives ESO2 in Mt; the constant 2 takes into account the difference in the gram-formula-weights of SO2 and S. Tables 1 and 2 provide data for the parameters, all of which are well-constrained, except V. W.E. Scott and others (this volume) report a range of 4-5 km3 DRE for V. Since the distal volume of tephra fall remains uncertain and may be too low, we will use the 5 km3-end of the range in this report. (All eruption product volumes in this report are DRE volumes.)
The usual practice for calculating Sm is to subtract the mean S concentration of the matrix glass from the mean S concentration of glass inclusions in a host phase (Sm= mean ppm Sinclusion, phase x - mean ppm Smatrix). For the June 15 Pinatubo dacites, however, there is no significant difference between the mean sulfur concentration of the matrix glasses and that of glass inclusions in any of the host phases (table 2, fig, 1). Statistical analysis of the data shows that for all glass inclusion hosts, Sm is not significantly different from zero at the 95 percent confidence level; indeed, it is not significantly different from zero at the 90 percent confidence level, except for glass inclusions in quartz. Thus, the petrologic method predicts an SO2 emission (ESO2) for the climactic June 15 eruption that is not significantly different from zero but is significantly different indeed from the 17-Mt remote sensing result.
PETROLOGIC EMISSION ESTIMATE FOR Cl
The petrologic emission estimate for Cl (ECl) is obtained from the Cl analog of equation (3). Since the melt-Cl loss, Clm (= mean ppm Clinclusion - mean ppm Clmatrix), is not significantly different from zero either (table 2, fig. 2), a zero petrologic emission estimate is also implied for Cl.
DISCUSSION OF ZERO PETROLOGIC EMISSION RESULTS FOR SO2 AND Cl
In making petrologic emission estimates for SO2 and Cl, we assume that the analyzed glass inclusions represent the sulfur and chlorine contents of the melt from which their host crystals precipitated, and that they retain this sulfur and chlorine contents after entrapment. The inclusions may have lost sulfur and chlorine, however, resulting in the zero values for Sm, Clm, ESO2, and ECl. Water-loss from glass (melt) inclusions by diffusive re-equilibration with external melt through quartz crystals is feasible (Qin and others, 1992); a similar process may cause sulfur- and chlorine-loss. Sulfur and chlorine could be lost by degassing if inclusions vesiculated due to crystallization, decompression, or thermal contraction after entrapment (Anderson, 1991; Skirius and others, 1990; Tait, 1992). Decompression during ascent and eruption may crack host crystals, also leading to loss of sulfur and chlorine (Tait, 1992).
We did not see textural evidence of glass crystallizing inside the inclusions nor at the glass-host crystal boundaries, although the latter is difficult to identify. Some glass inclusions are cracked, but they are a minority and restricted to quartz hosts, while rounded inclusions of clear glass with no visible cracks containing relatively large bubbles are common. Nearly all glass inclusions contain bubbles (Westrich and Gerlach, 1992), commonly with volumes larger than expected from melt shrinkage during cooling (~1-2 percent). Bubbles in hornblende-hosted glass inclusions have voids of up to 60 volume percent. Hornblende phenocrysts made poor glass inclusion containers, and they apparently underwent sufficient dilation stress upon decompression and subsequent vapor exsolution to deform along cleavage planes (Westrich and Gerlach, 1992). This explains the larger void volumes and significantly lower H2O contents of hornblende-hosted glass inclusions compared to inclusions in stronger containers like quartz phenocrysts (table 2). The S and Cl contents of glass inclusions in hornblende are, nevertheless, similar to the S and Cl contents of inclusions in other hosts, despite the clear differences in H2O contents (table 2). Perhaps the glass inclusions leaked sulfur and chlorine down to matrix glass-like concentrations the moment a bubble or crack was available. Because of their size and ubiquity, we suggested that the bubbles may represent a deep magmatic vapor phase present during entrapment (Westrich and Gerlach, 1992). Although this origin for the bubbles is difficult to confirm (Lowenstern and others, 1991; Lowenstern, 1993), bubbles of primary magmatic vapor within entrapped melt would presumably acquire sulfur and chlorine from degassing of the coexisting entrapped melt during syn- and post-eruptive decompression and cooling, thus facilitating sulfur- and chlorine-loss from the glass inclusion.
Loss of sulfur and chlorine from the glass inclusions may be the cause of the zero Sm and Clm and, thus, the zero petrologic emission estimates for SO2 and Cl. However, preliminary results from analyses of glass inclusions in plagioclase and quartz phenocrysts of air-fall deposits, which are expected to be less affected by post-eruption vesiculation because of more rapid quenching upon eruption (Skirius and others, 1990), show the same range of sulfur and chlorine contents as the present data set from pyroclastic flow deposits (H.R. Westrich, unpub. data, 1993). We stress, moreover, that the sulfur contents of the glass inclusions and matrix glasses (60-88 ppm, table 2) are virtually identical to experimentally determined sulfur solubility values (Carroll and Rutherford, 1987) for evolved, anhydrite-saturated, hydrous silicate melts at temperatures (800°C), pressures (200 MPa), and oxygen fugacities (>NNO+1) similar to those of the June 15 dacite before eruption (table 1). Indeed, recent experiments on anhydrite-saturated, hydrous June 15 dacite melts at T, P, and fO2 similar to the preeruption values (table 1) yield a sulfur solubility limit of ~60 +-20 ppm (Rutherford and Devine, this volume). Therefore, an alternative explanation of the zero Sm and ESO2 values is that sulfur did not degas significantly from the melt of the June 15 dacite during ascent and eruption. Consequently, both matrix glasses and glass inclusions have similar S contents, and there is no need to suggest significant loss of S from the glass inclusions. Similarly, the zero Clm may also reflect the lack of Cl degassing from melt during ascent and eruption. By this alternative view, matrix glasses and glass inclusions of the June 15 dacite behaved consistently: both show evidence of water loss during eruption, yet both appear to have lost insignificant S and Cl. It is unclear why this should be so, but perhaps the exsolution of S and Cl from rhyolite melt is much slower than water exsolution. The rapid decompression and dewatering of the melt (accompanied by a steep rise in viscosity) during ascent and eruption may have been too fast to permit reduction of melt sulfate and diffusion of SO2 to sites of growing water-rich bubbles (or, as the case may be, diffusion of sulfate to bubble-growth sites and reduction there to SO2). The lack of eruptive Cl degassing may also be related to exsolution kinetics; decrease in the diffusion rate of chlorine in rhyolitic melt as water exsolved may be a factor (Bai and Koster van Groos, 1994). Perhaps the decrease in the value of the vapor/melt distribution coefficient (DCl), as pressure drops, is involved (Shinohara and others, 1989; Webster and Holloway, 1990; Webster, 1992a).
It is unlikely that the glass inclusions ever contained enough sulfur to account for the 17-Mt SO2 cloud, however much sulfur they may or may not have lost. That would require a Sm of 1,344 ppm S for a V of 5 km3 (from equation (3) with ESO2 = 17 Mt). Adding the average matrix glass sulfur content of 60 ppm (table 2) implies ~1,400 ppm S for the glass inclusions before the conjectured sulfur loss. This concentration is not credible for the high-silica rhyolite melt phase of the dacite at preeruption conditions (table 1). It exceeds by over 23 times the ~60-ppm measured sulfur solubility of the June 15 dacite melts at conditions similar to those of the preeruption dacite (Rutherford and Devine, this volume).
PETROLOGIC EMISSION ESTIMATE FOR H2O
The difference between the water contents of the glass inclusions and matrix glasses is significant (table 2). Water clearly did degas from the melt during ascent and eruption. The water contents of glass inclusions in the various host phenocrysts are variable. The difference is greatest between glass inclusions in hornblende and quartz. As discussed above, the hornblende phenocrysts made relatively weak glass inclusion containers compared to quartz. Thus, glass inclusions in hornblende vesiculated and lost more water than the quartz-hosted inclusions when decompressed during ascent and eruption. We therefore take the highest water content measured for glass inclusions in quartz as the best estimate of the water contents prior to any volatile loss. The difference between this value (6.56 wt percent) and the water content of the matrix glasses (0.31 wt percent) gives an eruptive H2O-loss for the melt, (H2O)m, of 6.25 wt percent. This result together with the data in table 1 gives petrologic emission estimates for water of 395 Mt from the H2O analog to equation 3:
EH2O = 10-11 (H2O)mmmV (4)
PROBLEMATIC SOURCES
OF EXCESS SULFUR
Conventional petrologic emission estimates for SO2 assume that the sulfur dissolved prior to eruption in the silicate melt of the erupted magma, as represented by the S concentrations in glass inclusions, is the sole source of emitted SO2. The June 15 climactic event, however, produced a quantity of SO2 requiring an amount of sulfur ~23 times in excess of what could be dissolved in the volume of erupted melt at the preeruption T, P, and fO2. Thus, the 17-Mt SO2 cloud requires a major immediate source(s) of "excess sulfur" -- that is, sulfur in addition to that dissolved in the preeruption melt of the erupted dacite and available at the moment the eruption began. Several sources of excess sulfur possible at the time of the eruption can be envisioned; however, most are problematic.
MELT IN NON-ERUPTED DACITE
The degassing of melt in non-erupted dacite is a possible immediate source of excess sulfur. The volume, V, of non-erupted dacite required can be calculated from equation (3) constrained by the amount of S needed to make up the difference between the remote sensing and petrologic emission estimate for SO2 (17 Mt) and by data for m, m, and Sm (tables 1 and 2). Since Sm is not significantly different from zero for the erupted dacite, it is unlikely to be greater than zero for the non-erupted dacite. Thus, the required volume of non-erupted dacite is infinitely large. If we ignore the statistical significance of dispersion in the glass S data and use the nominal mean Sm (19 ppm, calculated from data in table 2) the implied volume of degassed non-erupted dacite is 350 km3. However, the magma reservoir under the summit area of Mount Pinatubo, as defined by low velocity regions, is only 40-90 km3 (Mori, Eberhart-Phillips, and Harlow, this volume); if low velocity regions extending out 5 km south-southeast of the summit are included, the reservoir is just 60-125 km3. Even assuming, for purposes of illustration, that all the glass inclusions leaked during/after eruption and that before leaking they contained 120 ppm S (twice the S solubility limit at preeruption conditions), the calculated volume of non-erupted dacite (110 km3) is still unrealistically large. We conclude that the degassing of melt in non-erupted dacite is not a viable source of excess sulfur for repairing the imbalance between remote sensing and petrologic emission estimates for SO2.
BASALTIC MAGMA
Basalt magma was injected and mixed with the dacite a short time before the eruption to form the June 7-12 hybrid andesite lava dome (with basaltic inclusions) and the June 12 hybrid andesite scoria (Pallister and others, 1992; this volume). It is thus a candidate for directly supplying excess sulfur for the 17-Mt SO2 cloud. We doubt, however, that direct degassing of excess sulfur from basalt magma was significant. Fragments of basalt mixed or commingled with dacite are absent in the June 15 eruption products. The occasional xenocrystic olivines scattered throughout the dacite apparently are from mixing with a small amount of basalt at an earlier time (Bernard and others, this volume; Pallister and others, this volume). It seems unlikely that basalt would be absent in the eruption deposits if it contributed most of the 17 Mt of SO2, which would require ~2 km3 of basalt even for a generous S release of 2,000 ppm. It is difficult to understand how basalt underlying the dacite or intruding and commingling with it could degas SO2 explosively in large quantities but supply none of the erupted magma. Furthermore, the ubiquitous presence of anhydrite throughout the large volume of erupted dacite indicates an enhanced fO2 since long before the latest basalt mixing event (Rutherford and Devine, this volume).
Although we reject basalt degassing as the direct and immediate source of excess sulfur for the SO2 cloud, we do not reject suggestions that basalt was the source of sulfur in the dacite on a longer time scale (Hattori, 1993; this volume; Matthews and others, 1992; Pallister and others, 1992; this volume). The recent injection and mixing of basalt with dacite, suggests the possibility of a long-term process involving migration of SO2 (and other gases) upward into the dacite from intruding and underplating basalt. The high oxygen fugacity of the dacite (table 1) is problematic in this regard (Westrich and Gerlach, 1992), although recent work shows that gases containing SO2 and released from basalt at 1200°C can become increasingly oxidizing with cooling (Gerlach, 1993a).
ANHYDRITE
The decomposition of anhydrite during eruption is a hypothesis often invoked as a source of excess sulfur (Devine and others, 1984; Sigurdsson, 1990). It seems unlikely, however, that anhydrite decomposition during degassing accompanying the ascent and eruption of the Pinatubo dacite provided a significant fraction of the 17-Mt SO2 cloud, despite suggestions to the contrary (Rutherford and Devine, 1991; this volume). This much SO2 would require the decomposition of an amount of anhydrite about equal to half that present in the erupted dacite (Westrich and Gerlach, 1992), as represented by 0.13-0.48 wt percent SO3 in whole-rock analyses (Bernard and others, 1991; this volume; Pallister and others, this volume). The implied decomposition of a substantial fraction of preeruption anhydrite is inconsistent with the appearance of anhydrite in the deposits.
Bernard and others (1991) report, "Most [anhydrite] crystals [in the dacite] are euhedral to subhedral with sharp contacts against the groundmass; no reaction coronae have been observed." And later: "The absence of reaction coronae, as well as the observation of frequent inclusions within anhydrite phenocrysts, strongly suggests that anhydrite was in equilibrium with the silicate melt at the time the magma erupted." Fournelle and others (this volume) found some anhydrites with rounded corners, possibly indicating decomposition. They comment that several anhydrite inclusions in plagioclase phenocrysts are fully rounded, although these surely formed before ascent and eruption. Pallister and others (this volume) note that anhydrite forms euhedral crystals in the June 15 dacite and the June 12 hybrid andesite scoria. But they describe anhydrite in the hybrid andesite of the June 7-12 dome, as highly resorbed anhedral masses with dusty reaction rims containing oxide and sulfide minerals, which they attribute to breakdown during slow cooling in degassed dome rocks -- not to breakdown during ascent and eruption. Nothing like this is seen in the more rapidly cooled and degassed dacite. Finally, a critical step in decomposing anhydrite to produce SO2 during ascent and eruption is the reduction of S6+ in anhydrite to S4+ in SO2. We know of no evidence for syn-eruptive oxidation of phases in the June 15 dacite.
The anhydrite breakdown hypothesis involves other problems as well. Over 2 hours are required for just 10 percent decomposition of anhydrite to CaO, SO2 and O2 in air at atmospheric pressure and 1,310°C (fig. 4) (Hanic and others, 1985). Furthermore, the decomposition rate decreases sharply with falling temperatures. The decomposition rate data indicate that anhydrite breakdown at atmospheric pressure and temperatures at or below the 780°C preeruption temperature of the Pinatubo dacite would be insignificant, perhaps even undetectable. This severely weakens the case for anhydrite decomposition to SO2 in the Pinatubo eruption cloud as a source of excess sulfur.
Figure 4. Rates for the anhydrite decomposition reaction
CaSO4(s) 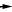 CaO(s) + SO2(g) + 1/2O2(g)
CaO(s) + SO2(g) + 1/2O2(g)
at various temperatures in air at atmospheric pressure. Data are from Hanic and others (1985). Over 2 hours are required for 10 percent decomposition at 1310°C. Decomposition times increase sharply at lower temperatures and thus suggest that anhydrite breakdown to SO2 in the Pinatubo eruption cloud is unlikely.
Experimental data suggest that coupled anhydrite breakdown and sulfate reduction could have occurred in a 3- to 6-hour decompression ascent of the Pinatubo dacite from depth (Baker and Rutherford, 1992; Rutherford and Devine, this volume). The dacite ascent times, however, were probably greatly less than an hour during the climactic eruption. Consider an erupting magma rising within a cylindrical conduit of radius, r, from a reservoir depth, d. Let v denote the average volume fraction of vapor, the crystal-melt density, V the DRE volume of erupted crystal-melt products, and te the duration of the eruption. The average mass flux, M, of crystal-melt products per unit cross-sectional conduit area is
M = V/te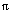 r2 (5)
r2 (5)
and the average rise velocity of the magma in the conduit is
v = M/(1-
v) (6)
(Jaupart and Allegre, 1991). The average conduit travel time, t
c, is thus
tc= d/v = r2dte(1-v)/V (7)
To maximize the calculated values of tc, v was assumed to be zero, and a larger than expected conduit diameter of 100 m was assumed. Observed conduit diameters are more like ~10-30 m (Chadwick and others, 1988; Swanson and Holcomb, 1989; Jaupart and Allegre, 1991). The initial sighting of the June 7-10 lava dome revealed a plug ~100 m across, suggesting a conduit diameter less than this amount (R. Hoblitt and C. Newhall, oral commun., September 22, 1993). The initial post-eruption dome had a diameter of only several tens of meters (C. Newhall, oral commun., September 22, 1993). The average depth to tapped magma, d, was taken to be 9 km (table 1). Estimates for the values of parameters te, V and are listed in table 1. The calculated conduit travel time is 7.6 minutes. The estimated travel times, moreover, are undoubtedly too long, because of the values assumed for r and v. Furthermore, since plinian activity and pyroclastic flow production seem to have occurred largely during the first 3 hours of the eruption (Wolfe and others, this volume), the 9-hour te value (table 1) may be too large by a factor of ~3. If so, conduit travel times could have been <150 s and in good agreement with travel times predicted from recent models for two-phase flow of gas-particle mixtures through eruption conduits (Sparks and others, 1994). If anhydrite decomposed this rapidly to produce the 17-Mt SO2 cloud, it is unclear why the anhydrite remaining in the pumice is predominantly as non-reacted euhedral and subhedral crystals. Perhaps the time needed for decompression breakdown of anhydrite could be satisfied if the erupted dacite decompressed and degassed in the magma chamber for 3 to 6 hours before ascent (Rutherford and Devine, this volume), but it is unclear how a decompression of the required magnitude (100-200 MPa) and duration could have occurred at 6-11 km; presumably such an event would also favor breakdown of cummingtonite and hornblende. Finally, we note that adiabtic cooling during rapid ascent (Wilson and others, 1980) would tend to enhance anhydrite stability.
HYDROTHERMAL FLUID
Another possible source of the 17-Mt SO2 discharge is the sulfate-rich fluid of the Mount Pinatubo hydrothermal system (Delfin and others, 1992; this volume). SO2 gas is not produced in significant amounts from dissolved sulfate when hydrothermal systems boil; it is nearly undetectable in the steam released from geothermal well discharges (Giggenbach, 1980). SO2 gas is unstable in the presence of hydrothermal liquid, relative to dissolved sulfate, which it tends to form by disproportionation reaction with water (Giggenbach, 1987; Williams and others, 1990). It is conceivable, however, that significant amounts of SO2 could be released if large amounts of sulfate-rich fluid came in contact with magma during the eruption and flashed to superheated vapor.
If we assume that the sulfate-rich samples from geothermal exploration wells (Delfin and others, 1992; this volume) are characteristic of the Pinatubo hydrothermal system, then hydrothermal fluids such as those observed in exploration well PIN-2D containing 1,129 ppm sulfate (table 3) give a bulk SO2 content of 753 ppm potentially available as an excess sulfur source. Even at this concentration level, at least 22,580 Mt of hydrothermal fluid, or ~23 km3 liquid-volume-equivalent (at STP), are required to produce 17 Mt of SO2, assuming all the sulfate is converted to SO2. We doubt that the heat required to flash this volume of hydrothermal fluid could have been transferred from 5 km3 of dacite within the 9 hours of June 15 eruption. Thermodynamic modeling of the flash-vaporization products indicates, moreover, that the potentially available SO2 would not be realized, even if 23 km3 of fluid did flash.
Table 3. Composition of a hydrothermal fluid sample from Pinatubo geothermal exploration well PIN-2D
[Data from Delfin and others (1992). Units are in ppm (wt). Sample collected 13 February 1989.]
|
Species
|
Concentration
|
|
pH
|
2.3 |
|
Na
|
1153 |
|
K
|
208 |
|
Ca
|
18 |
|
Mg
|
96 |
|
Li
|
2.4 |
|
Cl
|
1914 |
|
SO4
|
1129 |
|
B
|
67 |
|
SiO2
|
454 |
We used GASWORKS, a thermochemical equilibrium computer code (Symonds and Reed, 1993), to compute heterogeneous equilibrium distributions of gas, liquid, and mineral species as a function of temperature and pressure for complete flash-vaporizing of 22,580 Mt of Pinatubo hydrothermal fluid, with the composition of the PIN-2D sample (table 3), to superheated vapor. Since PIN-2D has a slight negative net charge, we subtracted small amounts of chloride, the most abundant anion, from the analysis to obtain charge balance in the starting fluid composition for the calculations. We neglected the small amounts of Li and B reported in the analysis (table 3). For the 9-element system, calculations included 117 gas species and 65 minerals. We began the flash-vaporization model by calculating the equilibrium distribution of gas and mineral species that form with complete instantaneous vaporization of the hydrothermal fluid to superheated vapor at 800°C. We then decreased temperature and recalculated the equilibrium distributions at 10°C decrements down to 400°C. In these calculations, the pressure was atmospheric (0.1 MPa), and the system was treated as a closed system -- no mineral fractionation or external buffering of components was allowed during cooling.
The quantities of gas and mineral species calculated in the equilibrium distributions at 800°C and 400°C are summarized in table 4. The results show that >99.9 percent of the hydrothermal sulfur precipitates as Na2SO4(s) and that very little sulfur remains in the gas phase as SO2 (<0.004 Mt). SO2 is the main sulfur gas species in the gas phase over the temperature range 800°C to 400°C, but its concentration is always very low (<=10-8 mole fraction), as shown in figure 5. Repeating the calculations at higher pressures up to the hydrostatic pressure of ~16 MPa expected at the 1,600 m bottom depth of the exploration well again showed that >99.9 percent of the hydrothermal sulfur precipitates as Na2SO4(s). Repeating the calculations for open system conditions allowing minerals to fractionate with cooling produced nearly identical abundances of SO2 to those of the closed system model.
Figure 5. Mole fraction of SO2 for closed system flash-vaporization of PIN-2D well fluid (table 3) from 800°C to 400°C at atmospheric pressure. Concentration of H2O is shown for reference. SO2 is the main sulfur gas species, but its concentration is always <10-8.
Table 4. Calculated equilibrium distributions of gas species and minerals for flash vaporization of a 22,580 Mt quantity of Pinatubo hydrothermal fluid containing 17 Mt bulk SO2.
[Based on hydrothermal fluid sample from geothermal well PIN-2D, table 3. Units are in Mt, megatons. Only SO2 and gas species more abundant than SO2 are shown. -, mineral not present at this temperature.]
|
Species
|
800°C
|
400°C
|
|
Gases
|
|
|
|
H2O
|
22,470 |
22,470 |
|
HCl
|
14.0 |
13.3 |
|
(NaCl)2
|
23.6 |
7x10-6 |
|
NaCl
|
11.5 |
5x10-6 |
|
KCl
|
7.9 |
4x10-6 |
|
O2
|
.9 |
0.8 |
|
(KCl)2
|
1.0 |
8x10-6 |
|
SO2
|
.004 |
6x10-9 |
|
Minerals
|
|
|
|
Na2SO4(1)
|
37.7 |
36.2 |
|
MgSiO3(2)
|
7.9 |
8.9 |
|
SiO2(3)
|
4.2 |
4.9 |
|
(Ca,Mg)SiO3(4)
|
2.2 |
- |
|
NaCl (halite)
|
- |
36.4 |
|
KCl (sylvite)
|
- |
8.9 |
|
CaSO4 (anhydrite)
|
- |
1.4 |
These calculations indicate it is not possible to produce 17 Mt of SO2 by flash-vaporization of Pinatubo hydrothermal fluid to superheated vapor. To get the 17 Mt of SO2 at 800°C, absurd quantities of ~105 km3 of fluid need to be flashed; at 400°C, the volume of fluid required balloons to >1010 km3! These results suggest further that a magmatic source -- not flash-vaporization of the hydrothermal system -- supplied the high SO2 emissions (up to 5,000-13,000 tons/day) observed in the weeks before the June 15 eruption (Daag, Tubianosa, and others, this volume).
PREERUPTION VAPOR SOURCE
OF EXCESS SULFUR
We show below that the dacite erupted on June 15 was vapor-saturated at depth prior to eruption, and propose that an accumulated vapor phase in the dacite provided the immediate source of excess sulfur for the 17-Mt SO2 cloud (Westrich and Gerlach, 1992; Gerlach, 1993b). As used here, "vapor" denotes a single volatile-rich phase having a distinctly lower density than that of the silicate liquid (melt) phase of the June 15 dacite. It is possible the dacite contained multiple (immiscible) volatile-rich phases, but lacking evidence to the contrary, we assume that a single volatile-rich phase predominated at depth. Fluid unmixing may have developed, however, during decompression ascent (Pasteris and others, this volume). It is also assumed that the vapor phase was dispersed as bubbles throughout the body of erupted dacite, although we recognize that the distribution may not have been uniform and that a vapor cap or foam layer (Jaupart and Vergniolle, 1989) may have existed near the top of the body.
VAPOR SATURATION
Low velocity regions defined by seismicity data (Mori, Eberhart-Phillips, and Harlow, this volume) permit a depth range of 6-11 km for the magma reservoir. Geothermal exploration and drilling data suggest that the volcano is underlain by about 2 km of non-lithified dacite (density=~2.0x1012
kg/km3) and 2 km of lithified dacite and intrusions (density=~2.6x1012 kg/km3), which overlie ophiolitic rocks (density=~2.9x1012 kg/km3) (Delfin, 1983; Pallister and others, this volume). We therefore assume an average crustal density of 2.7x1012 kg/km3, which gives a pressure range of ~160-300 MPa for the magma at 6-11 km in depth. This range is consistent with the preeruption pressure of 220+-50 MPa obtained by Al-in-hornblende geobarometry for the rims of hornblende phenocrysts (Rutherford and Devine, 1991; this volume); the uncertainty of +-50 MPa is the estimated error (M. Rutherford, oral commun., December, 8, 1993). These pressures also agree with experimentally determined stability limits for the phenocryst assemblage of the Pinatubo dacite at the preeruption temperature (780°C) (Rutherford and Devine, 1991; this volume). Thus, vapor saturation prior to eruption is plausible if vapor pressures significantly >160-170 MPa can be demonstrated.
The volatile contents of glass inclusions and the results of phase equilibria experiments are consistent with preeruption vapor saturation for the June 15 Pinatubo dacite (fig. 6). As discussed above, we take the highest water contents (6.1-6.6 wt percent) determined in this study for glass inclusions in quartz hosts as the best estimate for the water content of inclusions prior to any volatile loss. These water contents correspond to PH2O of ~210-250 MPa according to experimental solubility data for water in hydrous rhyolitic melt at the preeruption temperature (Silver and others, 1990). Rutherford and Devine (this volume) conclude from similar results that the water contents of glass inclusions correspond to PH2O of ~220 MPa for hydrous rhyolitic melt. They also report that water pressures of ~200-220 MPa are required to reproduce the composition of the melt in equilibrium with the natural phenocrysts of the June 15 dacite (Rutherford and Devine, this volume), and these pressures are presumably indicative of PH2O a short time before the eruption. The above values for PH2O are sufficient to make vapor saturation of the preeruption dacite plausible, even if water is assumed to have been the only volatile present. Thermodynamic constraints for C-O-H-S-volatile bearing magma require additional vapor pressure contributions, principally from CO2 and SO2. Rutherford and Devine (this volume) report that infrared spectroscopic analysis of glass inclusion-bearing quartz and plagioclase shows CO2 below detection (20 ppm). Wallace and Gerlach (1994), on the other hand, report infrared spectroscopic analyses that show 280-420 ppm CO2 for intact glass inclusions in quartz phenocrysts from splits of the samples used in this study. Their inclusions contain 6.1 to 6.6 wt percent water and ~70 ppm S. The water and carbon dioxide contents of these glass inclusions imply PH2O+ PCO2 of 250-290 MPa, based on the method of Newman and others (1988) (revised to include recent experimental data for H2O and CO2 solubility in rhyolitic melt (Silver and others, 1990; Blank and Stolper, 1993) and a modified Redlich-Kwong equation of state (Holloway, 1977) for modeling the properties of the vapor). The mean PH2O + PCO2 is 270 MPa. Experimental data indicate that anhydrite stability at the preeruption conditions would have required yet an additional vapor pressure contribution from PSO2, which, although uncertain at this time, is thought to be in the range 2-11 MPa (Baker and Rutherford, 1992; Rutherford and Devine, this volume). Finally, the 60-88 ppm S dissolved as sulfate in the June 15 glass inclusions and matrix glasses is compatible with a water-rich vapor phase. As noted above, experimental solubility data (Rutherford and Devine, this volume) show that the melt phase of June 15 dacite dissolves sulfur at this concentration level and oxidation state when saturated with anhydrite and water-rich vapor at the preeruption T, P, and fO2. Considering all the above results (fig. 6), we are persuaded that the total vapor pressure of the volatile components (mainly PH2O + PCO2 + PSO2) of the June 15 Pinatubo dacite was sufficient to cause saturation with H2O-rich vapor at depth in the magma reservoir prior to ascent and eruption.
Figure 6. Total pressure and vapor pressure estimates for the June 15 dacite. Permitted magma reservoir depth range is based on seismic velocity data (Mori, Eberhart-Phillips, and Harlow, this volume) and an estimated average crustal density of 2.7x1012 kg/km3, as discussed in text. Total pressure determined from the Al-in-hornblende geobarometer is 220 MPa with an estimated error of +-50 MPa (Rutherford and Devine, 1991; this volume). Several observations are consistent with vapor saturation and suggest vapor pressures in excess of 210 MPa. Water contents of 6.1-6.6 wt percent for glass inclusions in quartz hosts (Rutherford and Devine, this volume; this study) correspond to PH2O of ~210-250 MPa for water-rhyolitic melt systems at the preeruption temperature (Silver and others, 1990). Wallace and Gerlach (1994) report 280-420 ppm CO2 and 6.1 to 6.6 wt percent water for intact glass inclusions in quartz phenocrysts from splits of samples used in the present study, implying PH2O + PCO2 of 250-290 MPa (mean=270 MPa). Experimental studies (Rutherford and Devine, this volume) indicate that water pressures of ~200-220 MPa are required to reproduce the composition of the melt in equilibrium with the natural phenocrysts of the June 15 dacite (Rutherford and Devine, this volume), presumably indicative of PH2O a short time before the eruption. Other experimental data (Baker and Rutherford, 1992; Rutherford and Devine, this volume) indicate that anhydrite stability at the preeruption conditions would have required an additional vapor pressure contribution from PSO2 of 2-11 MPa; thus, experimental data suggest that the total vapor pressure (excluding PCO2) just prior to eruption was at least 202-231 MPa.
Exploration studies for geothermal energy at Mount Pinatubo strongly support the suggestion of vapor saturation and buildup in the dacite of the Pinatubo magma reservoir prior to the 1991 eruption. Three deep exploration wells were drilled into the Pinatubo hydrothermal system between August 1988 and November 1989. Samples and observations from these wells indicate "a significant magmatic input" into the hydrothermal system, and provide the basis for a model invoking magma as a source of volatiles observed in the hydrothermal fluids (Delfin and others, 1992; this volume; Ruaya and others, 1992). Chemical and stable isotope data show that the acidic hydrothermal fluids and gases encountered in the wells have strong similarities to high-temperature volcanic gases; these similarities suggest volatile discharge from the underlying magma (Casadevall, 1992; Delfin and others, 1992; this volume; Ruaya and others, 1992). Absorption of the magmatic volatile discharges in the hydrothermal system produced the acidic waters and generated the high sulfate fluids (table 3) (Delfin and others, 1992; this volume), presumably by disproportionation of magmatic SO2.
Finally, we speculate that the ubiquitous presence of bubbles in the Pinatubo glass inclusions (Westrich and Gerlach, 1992) supports a vapor saturation model. The pressure difference between entrapped melt and matrix melt is greater during decompression if the entrapped melt was vapor-saturated (Tait, 1992). Thus, entrapped vapor-saturated melt would have a greater tendency to deform host crystals and vesiculate during eruption. This would promote deformation of phenocrysts and the development of glass inclusions with relatively large bubble volumes, as commonly observed in the June 15 dacite (Westrich and Gerlach, 1992).
VAPOR COMPOSITION
We attempt to model the approximate composition of the preeruption vapor present a short time before the climactic eruption. The model is constrained by the mass abundance of SO2 in the vapor phase and constraints on the vapor pressures. The mass abundance of SO2 in the vapor phase is assumed to be 17 Mt, based on the remote sensing data, which is regarded as a minimum estimate, as discussed previously. PCO2 is taken to be 40 MPa, based on experimental solubility measurements for rhyolitic liquids and the dissolved CO2 content of glass inclusions (Wallace and Gerlach, 1994). We have used a water pressure of 220 MPa to be consistent with experimental PH2O values that reproduce the composition of the melt in equilibrium with the natural phenocrysts of the June 15 dacite (Rutherford and Devine, this volume), and that presumably reflect the vapor composition a short time before the eruption. The value of PSO2 a short time before eruption is not as well constrained. PSO2 values used here are based on experimental data for anhydrite stability in the June 15 dacite (Baker and Rutherford, 1992). The initial analyses of experimental run products indicated a PSO2 of ~11 MPa, which leads to a total (model) pressure of ~275 MPa (including PCl, as discussed below). Recent analyses suggest a PSO2 of ~2 Mpa and give a total (model) pressure of ~265 MPa. Both values for PSO2 are used in the model calculations that follow, however we have reservations about the results constrained by the 2-MPa PSO2 value, as discussed below.
The molar abundances of H2O and CO2 in the vapor phase are calculated from simple equations of the form
Ni = Pi(NSO2/PSO2) (8)
where Ni is the molar abundance of species i in the vapor phase. This equation links intensive parameters (PSO2) evaluated from petrologic experiments and extensive parameters (NSO2) obtained from remote sensing measurements. The resulting molar and mass abundances are given in table 5 and indicate a H2O-rich vapor. Experimental studies of natural and synthetic rhyolites at Pinatubo-like conditions of 800°C and 200 MPa show that Cl partitions preferentially into aqueous vapor relative to melt (Webster and Holloway, 1990; Webster, 1992a, b; Metrich and Rutherford, 1992). These studies suggest that for a preeruption melt Cl content of 0.102 wt percent in the Pinatubo dacite, based on the average Cl content of the glasses (table 2), the vapor-melt distribution coefficient for Cl, DCl, given by
DCl = (wt percent Clvapor)/(wt percent Clmelt) (9)
is roughly 20, indicating ~2 wt percent Cl in the vapor phase. Estimated total Cl abundances in the vapor (mainly as NaCl, KCl, and HCl) are included in table 5.
The composition of the inferred preeruption vapor is summarized in table 5. The mole percent results for the upper- and lower-PSO2
values are similar. The vapor phase was not excessively SO2-rich. It contained only ~4 mole percent or less SO2 despite the impressive 17-Mt SO2 emission. The vapor also contained ~80-83 mole percent H2O, ~15 mole percent CO2, and ~1 mole percent Cl (table 5). We stress that the molar and mass abundances of H2O, CO2, and Cl given in table 5 are minimum values, since they are linked to the minimum emission estimate of 17 Mt for SO2.
The concentration of sulfur obtained for the preeruption vapor compared to its concentration in the melt indicates a high volatility for sulfur in the June 15 dacite. The distribution coefficient, DS, for sulfur between the preeruption vapor and melt of the dacite can be calculated from an analog to equation (9), and provides a measure of sulfur volatility. The wt percent concentration of S in the preeruption vapor is equal to half that of SO2 (table 5). The preeruption S content of the melt is taken to be 0.0075 wt percent, based on the average S content of the glasses (table 2). Thus, DS is ~720 (PSO2 = 11 MPa) or ~140 (PSO2 = 2 MPa), underscoring a strong preference for sulfur to reside in the vapor as SO2 compared to dissolution in the melt as sulfate, despite the highly oxidizing preeruption conditions.
Table 5. Composition of the preeruption vapor present in the dacite of the climactic June 15, 1991, eruption, Mount Pinatubo.
[Mt, megaton; Tmol, teramole (1012); MPa, megapascal]
|
Component
|
Mass1
(Mt)
|
Moles1
(Tmol)
|
Mole Percent
|
Pressure
(MPa)
|
|
PSO2= 11 MPa
|
|
|
|
|
|
H2O
|
95.6 |
5.3073 |
80.1 |
220 |
|
SO2
|
17. |
0.2654 |
4.0 |
11 |
|
CO2
|
42.5 |
0.9650 |
14.5 |
40 |
|
Cl
|
3.2 |
0.0911 |
1.4 |
3.8 |
|
Totals :
|
158.3 |
6.6288 |
100.0 |
274.8 |
|
PSO2= 2 MPa
|
|
|
|
|
|
H2O
|
525.9 |
29.1901 |
82.9 |
220 |
|
SO2
|
17. |
0.2654 |
0.8 |
2 |
|
CO2
|
233.6 |
5.3073 |
15.1 |
40 |
|
Cl
|
16.1 |
0.4560 |
1.2 |
3.4 |
|
Totals :
|
792.6 |
35.2188 |
100.0 |
265.4 |
VAPOR VOLUME AND DENSITY
The ideal gas law gives an estimate of the approximate total volume of vapor at depth, Vvapor, directly from the 17-Mt SO2 emission (NSO2 = 0.2654x1012 moles, table 5), the preeruption PSO2 (2 and 11 MPa), and T780°C), as follows:
Vvapor = (N/P)RT = (NSO2/PSO2)RT (10)
The results are 0.21 km3 (PSO2 = 11 Mpa) and 1.2 km3 (PSO2 = 2 MPa). The corresponding ideal gas density estimates for the vapor masses given in table 5 are 0.75 g/cm3 (PSO2 = 11 MPa) and 0.68 g/cm3 (PSO2= 2 MPa).
An ideal mixing model for the vapor volume, based on endmember volumes obtained from PVT data for H2O (Burnham and others, 1969) and from a modified Redlich-Kwong equation of state (Holloway, 1977, 1981) for CO2 and SO2, gives Vvapor of 0.25 km3 (PSO2 = 11 MPa) and 1.35 km3 (PSO2 = 2 MPa). (The negligible Cl vapor was treated as ideal gas.) The respective density estimates are 0.63 g/cm3 and 0.59 g/cm3.
The vapor phase volume results are relatively insensitive to total pressure and vapor abundance. This is because ideal and nonideal values for Vvapor tend to be a strong function of the quantities N/P and RT, which are fixed by geothermometry data for T (780°C, table 1) and by remote sensing data and anhydrite stability data for NSO2/PSO2
(for example, 0.2413x1011 mole/MPa for PSO2 = 11MPa). As a result, changes in pressure (P) and vapor abundance (N) are coupled such that N/P remains fixed, which tends to stabilize Vvapor.
DACITE VAPOR FRACTION AND
DENSITY BEFORE ERUPTION
We estimate the volume fraction of vapor at depth in the preeruption dacite from the ideal mixing vapor volume (above) and 5 km3 of erupted dacite. Figure 7 shows the results expressed as volume percent vapor in the preeruption dacite at 780°C and pressures of 265-275 MPa; the pressure range corresponds to the PSO2 values of 2 and 11 MPa. The vapor fraction ranges from 4.8 volume percent (PSO2 = 11 MPa) to 21 volume percent (PSO2 = 2 MPa). The vapor fraction increases sharply for PSO2 less than 5 MPa.
Figure 7. Volume percent vapor at depth in the preeruption dacite. The curve is for vapor compositions at 780°C from PSO2 = 2 MPa (P = 265 MPa) to PSO2 = 11 MPa (P = 275 MPa). The results assume ideal mixing in the vapor of end member volumes that are based on pressure-volume temperature data for H2O (Burnham and others, 1969) and a modified Redlich-Kwong equation of state for CO2 and SO2 (Holloway, 1977, 1981).
The volume fractions shown in figure 7 may be minimum estimates because of their dependence on the minimum SO2 emission estimate of 17 Mt. Nevertheless, it is possible they may be too high, since it is assumed that all vapor was derived only from the erupted dacite. Although derivation of vapor by syn-eruptive degassing of melt in non-erupted dacite was apparently insignificant, as discussed above, our analysis does not rule out syn-eruptive escape of the preeruption vapor phase from non-erupted (vapor-saturated) dacite. It seems unlikely to us, however, that large amounts of such vapor could segregate and escape from non-erupted dacite fast enough to supply the explosive eruption without simultaneous ejection of the source material.
The preeruption bulk density of the dacite can be estimated from the sum of the vapor mass (table 5) and the erupted mass of crystals and glass (from data in table 1) divided by the sum of the ideal mixing vapor volume (Vvapor) and the DRE volume of erupted dacite. The results (fig. 8) range from 2.31x1012 kg/km3 (PSO2 = 11 MPa) to 2.01x1012 kg/km3 (PSO2 = 2 MPa).
Figure 8. Bulk density of the preeruption dacite. (Note: 1012 kg/km3 = 1 g/cm3.) The solid curve is for vapor compositions at 780°C from PSO2 = 2 MPa (P = 265 MPa) to PSO2 = 11 MPa (P = 275 MPa). Densities are calculated from the sum of the vapor mass (table 5) and the erupted mass of crystals and glass (from data in table 1) divided by the sum of the ideal mixing vapor volume (Vvapor) and the DRE volume of erupted dacite (table 1). Dotted line is the estimated density of the dacite without vapor, based on density estimates for vesicle-free dacite (table 1).
VAPOR ACCUMULATION
It is impossible to dissolve the total amount of volatiles contained in the preeruption vapor and melt in a completely molten, 5 km3 quantity of dacite at the magma reservoir pressures (<=300 MPa, fig. 6). Consider the mass of water, MH2O, dissolved in the rhyolite melt of the dacite and given by
MH2O = 10-11 (H2O)m m m V (11)
where MH2O is in Mt, (H2O)m is the wt percent water dissolved in the rhyolite melt; the other terms are defined as for equation 4 above and have values given in table 1. On the basis of a preeruption melt concentration of 6.56 wt percent water (table 2), MH2O is 415 Mt. Including the 95.6 Mt of water in the preeruption vapor based on PSO2 of 11 MPa (table 5) implies bulk water contents for completely molten dacite of 4.2 wt percent. Similarly, the estimated 42.5 Mt CO2 in the preeruption vapor (table 5) together with ~0.03 wt percent dissolved CO2 (Wallace and Gerlach, 1994) imply bulk carbon dioxide contents for completely molten dacite of 0.36 wt percent. Calculations based on the revised method of Newman and others (1988), indicate that to dissolve these amounts of water and carbon dioxide in rhyolite would require pressures >400 MPa. A vapor phase composition based on PSO2 of 2 MPa (table 5) requires the dissolution of 7.4 wt percent H2O and 1.8 wt percent CO2, which would necessitate still higher pressures. Thus, it is unlikely that the total amount of water, carbon dioxide, and sulfur dioxide could have been dissolved in the erupted quantity of dacite at reservoir pressures, even if completely molten, since the solubilities of water and carbon dioxide used here are for rhyolite and would decrease in higher temperature, less evolved dacitic melts (Holloway, 1981; Fogel and Rutherford, 1990; Pan and others, 1991). The effect of SO2 on the vapor saturation pressure, which has been neglected in this analysis, would presumably further increase the pressure required for complete volatile dissolution. Finally, the pressures required to achieve dissolution are very sensitive to the amounts of CO2, which are minimal in the above calculations because of linkage to the minimum emission estimate of 17 Mt for SO2.
The total volatile load contained in the vapor and dissolved in the melt of the erupted June 15 dacite apparently requires some process(es) of vapor accumulation either by open system volatile migration or by closed system crystallization/decompression (Wallace and Gerlach, 1994). We suggest that long term, open system migration of vapor upward from subjacent dacite and (or) mafic magmas intruding and underplating the magma chamber may have provided the main supply of excess volatiles for the accumulated vapor in the magma that erupted on June 15. Alternatively, the volatile load may have been dissolved in higher temperature, less crystallized dacite at depths greater than 11 km, with subsequent exsolution and accumulation as vapor during crystallization and decompression associated with cooling and emplacement at 6-11 km. If this were the case, however, at least some glass inclusions would be expected with more primitive and sulfur-rich compositions than are observed.
VOLATILE EMISSION ESTIMATES
FOR THE CLIMACTIC ERUPTION
The climactic eruption released the accumulated vapor of the erupted dacite and resulted in emissions of 17 Mt SO2, 96-526 Mt H2O, 42-234 Mt CO2, and 3-16 Mt of Cl (table 5). Additional yields of SO2 and Cl from degassing of melt were minor to insignificant during ascent and eruption. Degassing of melt during ascent and eruption may have released an additional 2 Mt CO2, depending on the kinetics of CO2 exsolution. Melt degassing of water was significant during ascent and eruption, and yielded an additional 395 Mt of H2O. Thus, the total H2O emission was approximately 491 Mt (PSO2= 11 MPa) to 921 Mt (PSO2= 2 MPa). The total volatile emission for the climactic eruption was 555-1190 Mt, of which 90 to 95 mole percent was water.
DISCUSSION AND CONCLUSIONS
We have more confidence in the vapor composition model constrained by a PSO2 of 11 MPa than in the model based on a PSO2 of 2 MPa (table 5). The latter model implies a C/S -- that is, the molar ratio of total C (CO2+CO+. . .) to total S (SO2+H2S+2S2+. . . ) -- for the vapor of ~20, compared to a C/S of ~3.6 in the former model. C/S values of high-temperature volcanic gases (>500°C) collected from active convergent plate volcanoes, however, are <20, except for a few samples collected from degassed domes (e.g., Showa-Shinzan) several years after dome emplacement (Marty and Le Cloarec, 1992; Williams and others, 1992; Symonds and others, 1994). A recent compilation of volcanic gas data (Symonds and others (1994) shows C/S values <20 for volcanic gases from volcanoes of all tectonic environments; the mean of the average C/S values for active convergent plate volcanoes is 6 with a standard error (s/n1/2) of 2. Thus, the C/S of ~20 implied by the 2-MPa PSO2 vapor composition model is unusually S-depleted, which seems unlikely for a magma that yielded 17 Mt of SO2, primary anhydrite, and 1,300-4,800 ppm SO3 in whole-rock analyses.
Several other implications of the 2-MPa PSO2 vapor composition model also give us reservations. These include (1) vapor fractions for the preeruption dacite of up to 21 volume percent, (2) preeruption dacite bulk densities as low as 2x1012 kg/km3, (3) bulk water contents up to 7.4 wt percent, and (4) total volatile emissions for the climactic eruption of up to 1,200 Mt. It is unclear how the dacite could acquire such high vapor fractions and low bulk densities without having erupted long ago, although recent work suggests similar vapor fractions and densities for the Bishop Tuff prior to eruption (Wallace and Anderson, 1994). In any event, this study underscores the need to examine the potential importance of a significant preeruption vapor fraction to the dynamics of explosive eruptions. Current models of the dynamics of magma degassing and explosive eruptions typically assume an initial condition of volatile undersaturation or of volatile saturation with only a few bubbles present (Sparks and others, 1994).
We conclude that the dacite magma erupted in the climactic June 15, 1991, eruption of Mount Pinatubo was vapor-saturated and contained an accumulation of at least 5 volume percent, water-rich vapor (~80 mole percent H2O) at depth prior to eruption. The accumulated vapor made the dacite buoyant and able to respond rapidly to decompression, thus increasing its "eruptability" and sensitivity to triggering mechanisms. The accumulated vapor was the chief immediate source of excess sulfur for the giant 17-Mt SO2 cloud injected into the stratosphere, although it was not excessively SO2-rich, containing only ~4 mole percent or less SO2. Other potentially available immediate sources of excess sulfur (non-erupted dacite, mafic magma, anhydrite decomposition, sulfate-rich hydrothermal fluids, and degassing of ascending melt) made relatively unimportant contributions to the 17-Mt SO2 cloud. Accumulated preeruption vapor also contributed emissions of at least 96 Mt of H2O, 42 Mt CO2 and 3 Mt Cl during the climactic eruption. Melt degassing of water was significant during ascent and eruption and yielded an additional 395 Mt of H2O, which undoubtedly played an important role in the dynamics of the eruption.
Conventional petrologic estimates based on glass inclusions predict statistically insignificant emissions for both SO2 and Cl during the climactic eruption. The low petrologic estimates for these volatiles result from their relatively high volatility and enrichment in the vapor phase of the dacite prior to eruption, coupled with apparently negligible degassing of SO2 and Cl from melt during ascent and eruption of the June 15 dacite. This study reinforces growing evidence, based on smaller eruptions (VEI <=5), that petrologic emission estimates for SO2 are many times lower than estimates based on remote sensing, and indicates that petrologic estimates may also seriously underestimate the SO2 emissions of larger explosive eruptions. If explosive volcanism commonly involves vapor-saturated magma containing accumulated vapor, petrologic estimates for SO2 emissions during explosive eruptions of the past may be far too low and significantly underestimate their impacts on climate and the chemistry of the atmosphere. Thus, an improved technique is sorely needed to infer the SO2 yields and potential global impacts of pre-TOMS eruptions.
Conventional petrologic emission estimates for Cl and CO2 probably are significantly low also, since these volatiles partition strongly into a vapor phase, while those for H2O may be only marginally low. Because it cannot be assumed that volatiles will invariably accumulate in preeruption melt like most other incompatible elements, the volatile contents of glass inclusions alone do not, in general, provide a sufficient basis for predicting either the total preeruption volatile contents of magma or the volatile emissions of explosive eruptions. When volatile determinations on glass inclusions are combined with volatile measurements on emission clouds, however, the results are complementary and provide improved constraints for the preeruptive volatile contents of magmas, including magmas containing large quantities of accumulated vapor. This study illustrates the potential utility of obtaining values for both intensive parameters (for example, T, P, PH2O, PCO2, PSO2) from petrologic measurements and extensive parameters (for example, NSO2) from emission measurements to improve estimates of the preeruption volatile contents of explosive magma.
We stress the need to make greater use of remote sensing techniques to measure volcanic emissions and to infer subsurface magmatic conditions. Applications of techniques for measuring CO2 emissions during eruptions, ideally in conjunction with glass inclusion studies, are especially encouraged. Because of its low solubility in silicate melts and the usual absence of carbonate minerals in magma, CO2 should be strongly enriched in the vapor phase of vapor-saturated magmas (Holloway, 1976). Thus, measurements of CO2 emissions in future eruptions, together with solubility data, can provide critical tests of the vapor saturation and accumulation model (see Gerlach and others, 1994).
Finally, we note that the apparent large distribution coefficient for sulfur between preeruption vapor and melt of the Pinatubo dacite (DS >100) suggests the importance of measuring DS experimentally for vapor-saturated dacitic and rhyolitic melts at a range of temperatures, pressures, and oxygen fugacities. Measurement of DS at appropriate experimental conditions would directly test the validity of our proposal that excess sulfur for the 17-Mt stratospheric SO2 cloud resided in a vapor phase at depth in the Pinatubo dacite prior to the climactic eruption.
ACKNOWLEDGMENTS
We are indebted to many people who helped us in the course of this study. Rick Hoblitt, John Pallister, and Ed Wolfe provided samples, and Rick Hervig at the Arizona State University's Center for Solid State Sciences assisted with ion probe analyses. Sally Newman provided calculations for estimating vapor pressures of rhyolitic melts containing carbon dioxide and water. Several authors of papers in this volume generously supplied their draft and revised versions of manuscripts containing critical data for our use. In this regard, we especially thank Alain Bernard, Joseph Devine, John Pallister, Malcolm Rutherford, Willie Scott, and Ed Wolfe. We are grateful to numerous colleagues who shared ideas with us, and we benefited greatly from our conversations with Gregg Bluth, Arlin Krueger, Jim Luhr, John Pallister, Bill Rose, Malcolm Rutherford, Steve Self, and Paul Wallace. We thank our reviewers Fred Anderson, Michael Carroll, Chris Newhall, Bill Melson, Haraldur Sigurdsson, and Ed Wolfe for prompt and careful reviews. Fred Anderson's comments were especially helpful in improving the conceptual framework of the manuscript. Finally, we wish to acknowledge John Watson of the USGS Branch of Eastern Technical Reports for his skill, enthusiasm, and patience in carrying out several manuscript revisions. The USGS Global Change and Climate History Program and the U.S. Department of Energy BES Geosciences Research Program funded the investigation.
REFERENCES CITED
Anderson, A.T., 1991, Hourglass inclusions: Theory and application to the Bishop Rhyolitic Tuff: American Mineralogist, v. 76, p. 530-547.
Andres, R.J., Rose, W.I., Kyle, P.R., deSilva, S., Francis, P., Gardeweg, M., and Moreno Roa, H., 1991, Excessive sulfur dioxide emissions from Chilean volcanoes: Journal of Volcanology and Geothermal Research, v. 46, p. 323-329.
Bai, T. B., and Koster van Groos, A. F., 1994, Diffusion of chlorine in granitic melts: Geochimica et Cosmochimica Acta, v. 58, p. 113-123.
Baker, L., and Rutherford, M. J., 1992, Anhydrite breakdown as a possible source of excess sulfur in the 1991 Mount Pinatubo eruption [abs.]: Eos, Transactions, American Geophysical Union, v. 73, p. 625.
Bernard, A., Demaiffe, D., Mattelli, N., and Punongbayan, R. S., 1991, Anhydrite-bearing pumices from Mount Pinatubo: Further evidence for sulfur-rich silicic magmas: Nature, v. 354, p. 139-140.
Bernard, A., Knittel, U., Weber, B., Weis, D., Albrecht, A., Hattori, K., and Oles, D., this volume, Petrology and geochemistry of the 1991 eruption products of Mount Pinatubo (Luzon, Philippines).
Bence, A.E., and Albee, A.L., 1968, Empirical correction factors for the electron microanalysis of silicates and oxides: Journal of Geology, v. 76, p. 382-403.
Blank, J. G., Stolper, E. M., 1993, Solubilities of carbon dioxide and water in rhyolitic melt at 850
C and 750 bars: Earth and Planetary Science Letters, v. 119, p. 27-36.
Bluth, G. J. S., Doiron, S. D., Schnetzler, C. C., Krueger, A. J., and Walter, L. S., 1992, Global tracking of the SO2 clouds from the June, 1991 Mount Pinatubo eruptions: Geophysical Research Letters, v. 19, p. 151-154.
Bluth, G. J. S., Schnetzler, C. C., Krueger, A. J., and Walter, L. S., 1993, The contribution of explosive volcanism to global atmospheric sulfur dioxide concentrations: Nature, v. 366, p. 327-329.
Burnham, W. C., Holloway, J., R., and Davis, N. F., 1969, Thermodynamic properties of water to 1,000°C and 10,000 bars: The Geological Society of America, Special Paper Number 132, p. 1-96.
Carroll, M.R., and Rutherford, M. J., 1987, The stability of igneous anhydrite: Experimental results and implications for sulfur behavior in the 1982 El Chichón trachyandesite and other evolved magmas: Journal of Petrology, v. 28, p. 781-801.
------1988, Sulfur speciation in hydrous experimental glasses of varying oxidation state: Results from measured wavelength shifts of sulfur X-rays: American Mineralogist, v. 73, p. 845-849.
Casadevall, T. J., 1992, Preeruption hydrothermal systems at Pinatubo, Philippines and El Chichón, Mexico: Evidence for degassing magmas beneath dormant volcanoes: Geological Survey of Japan Report, no. 279, p. 35-38.
Chadwick, W., Jr., Archuleta, R., and Swanson, D. A., 1988, The mechanics of ground deformation precursory to dome-building extrusions at Mount St. Helens 1981-1982: Journal of Geophysical Research, v. 93, no. B5, p. 4351-4366.
Chase, M. W., Davies, C. A., Downey, J. R., Frurip, D. J., McDonald, R. A., and Syverud, A. N., 1985, JANAF thermochemical tables: Journal of Physical and Chemical Reference Data, v. 14, Supplement no. 1, p. 1-1856.
Daag, A., Tubianosa, B., Newhall, C., Tungol, N., Javier, D., Dolan, M., Delos Reyes, P. J., Arboleda, R., Martinez, M., and Regalado, M. T. M., this volume, Monitoring sulfur dioxide emissions at Mount Pinatubo volcano.
Delfin, F. G., 1983, Geology of the Mount Pinatubo geothermal prospect: unpublished Philippine National Oil Company report, 35 p.
Delfin, F. G., Sussman, D., Ruaya, J. R., and Reyes, A. G., 1992, Hazard assessment of the Pinatubo volcanic-geothermal system: Clues prior to the June 15, 1991 eruption: Transactions Geothermal Resources Council, v. 16, p. 519-528.
Delfin, F. G., Villarosa, H. G., Layugan, D. B., Clemente, V. C., Candelaria, M. R., and Ruaya, J. R., this volume, Geothermal exploration of the pre-1991 Pinatubo hydrothermal system.
Devine, J. D., Sigurdsson, H., Davis, A. N., and Self, S., 1984, Estimates of sulfur and chlorine yield to the atmosphere from volcanic eruptions and potential climatic effects: Journal of Geophysical Research, v. 89, p. 6309-6325.
Dutton, E. G., and Christy, J. R., 1992, Solar radiative forcing at selected locations and evidence for global lower tropospheric cooling following the eruptions of El Chichón and Pinatubo: Geophysical Research Letters, v. 19, p. 2313-2316.
Fogel, R. A., and Rutherford, M. J., 1990, The solubility of carbon dioxide in rhyolitic melts: A quantitative FTIR study: American Mineralogist, v. 75, p. 1311-1326.
Fournelle, J., Carmody, R., and Daag, A. G., this volume, Mineraolgy and geochemistry of Mount Pinatubo anhydrite- and sulfide-bearing pumices from the SO2-rich eruption of June 1991.
Gerlach, T. M., 1993a, Oxygen buffering of Kilauea volcanic gases and the oxygen fugacity of Kilauea basalt: Geochimica et Cosmochimica Acta, v. 57, p. 795-814.
------1993b, A magmatic vapor saturation and accumulation model for the 20-Megaton SO2 cloud from the June 15, 1991, climactic eruption of Mount Pinatubo [abs.]: Eos, Transactions, American Geophysical Union, v. 74, no. 43, p. 104.
Gerlach, T. M., Westrich, H. R., Casadevall, T. J., and Finnegan, D. L., 1994, Vapor saturation and accumulation in magmas of the 1989-1990 eruption of Redoubt Volcano, Alaska: Journal of Volcanology and Geothermal Research, v. 62, p. 317-337.
Giggenbach, W. F., 1980, Geothermal gas equilibria: Geochimica et Cosmochimica Acta, v.44, p. 2021-2032.
------1987, Redox processes governing the chemistry of fumarolic gas discharges from White Island, New Zealand: Applied Geochemistry, v. 2, p. 143-161.
Gleason, J. F., Bhartia, P. K., Herman, J. R., McPeters, R., Newman, P., Stolarski, R. S., Flynn, L., Labow, G., Larko, D., Seftor, C., Wellemeyer, C., Komhyr, W. D., Miller, A. J., and Planet, W., 1993, Record low global ozone: Science, v. 260, p. 523-526.
Halpert, M. S., Ropelewski, C. F., Karl, T. R., Angell, J. K., Stowe, L. L., Heim, R. R., Jr., Miller, A. J., and Rodenhuis, D. R., 1993, 1992 brings return to moderate global temperatures: Eos, Transactions, American Geophysical Union, v. 74, no. 38, p. 433, 437-439.
Hanic, F., Galikova, L., Havlica, J., Kapralik, I., and Ambruz, V., 1985, Kinetics of the thermal decomposition of CaSO4 in air: British Ceramics Transactions Journal, v. 84, p. 22-25.
Hattori, K., 1993, High-sulfur magma, a product of fluid discharge from underlying mafic magma: Evidence from Mount Pinatubo, Philippines: Geology, v. 21, p. 1083-1086.
Hattori, K., this volume, Occurrence and origin of sulfide and sulfate in the 1991 Pinatubo eruption products.
Hervig, R. L., and Williams, P., 1988, SIMS microanalysis of minerals and glasses for H and D, in Huber, A. M., and Werner, A. W., eds., Secondary ion mass spectrometry, SIMS IV: New York, John Wiley & Sons, p. 961-964.
Hoblitt, R. P., Wolfe, E. W., Scott, W. E., Couchman, M.R., Pallister, J., and Javier, D., this volume, The preparoxysmal eruptions, Mount Pinatubo, Philippines.
Hofman, D. J., Oltmans, S. J., Harris, J. M., Deshler, T., and Johnson, B. J., 1992, Observation and possible causes of new ozone depletion in Antarctica in 1991: Nature, v. 359, p. 283-287.
Holloway, J. R., 1976, Fluids in the evolution of granitic magma: Consequences of finite CO2 solubility: Geological Society of America Bulletin, v. 87, p. 1513-1518.
Holloway, J. R., 1977, Fugacity and activity of molecular species in supercritical fluids, in Fraser, D., ed., Thermodynamics in geology: Boston, D. Reidel, p. 161-181.
Holloway, J. R., 1981, Volatile interactions in magmas, in Newton, R. C., Navrotsky, A., and Wood B. J., eds., Thermodynamic of Minerals and Melts: Springer-Verlag, p. 273-293.
Huebner, J. S., and Sato, M., 1970, The oxygen fugacity-temperature relationships of manganese oxide and nickel oxide buffers: American Mineralogist, v. 55, p. 934-952.
Imai, A., Listanco, E. L., and Fujii, T., 1993, Petrologic and sulfur isotopic significance of highly oxidized and sulfur-rich magma at Mt. Pinatubo, Philippines: Geology, v. 21, p. 699-702.
Jaupart, C., and Allegre, C. J., 1991, Gas content, eruption rate and instabilities of eruption regime in silicic volcanoes: Earth and Planetary Science Letters, v. 102, p. 413-429.
Jaupart, C., and Vergniolle, S., 1989, The generation and collapse of a foam layer at the roof of a basaltic magma chamber: Journal of Fluid Mechanics, v. 203, p. 347-380.
Johnston, D. A., 1980, Volcanic contribution of chlorine to the stratosphere: More significant to ozone than previously estimated?: Science, v. 209, p. 491-493.
Lowenstern, J. B., 1993, Evidence for a copper-bearing fluid in magma erupted at the Valley of Ten Thousand Smokes, Alaska: Contributions to Mineralogy and Petrology, v. 114, p. 409-421.
Lowenstern, J. B., Mahood, G. A., Rivers, M. L., and Sutton, S. R., 1991, Evidence for extreme partitioning of copper into magmatic vapor: Science, v. 252, p. 1405-1409.
Luhr, J. F., Carmichael, I. S., and Varekamp, J. C., 1984, The 1982 eruption of El Chichón volcano, Chiapas, Mexico: Mineralogy and petrology of the anhydrite-bearing pumices: Journal of Volcanology and Geothermal Research, v. 23, p. 69-108.
Marty, B., and Le Cloarec, M-F., 1992, Helium-3 and CO2 fluxes from subaerial volcanoes estimated from polonium-210 emissions: Journal of Volcanology and Geothermal Research, v. 53, p. 67-72.
Matthews, S. J., Jones, A. P., and Bristow, C. S., 1992, A simple magma-mixing model for sulphur behaviour in calc-alkaline volcanic rocks: mineralogical evidence from Mount Pinatubo 1991 eruption: Journal of the Geological Society, London, v. 149, p. 863-866.
McCormick, P.M., Thomason, L.W., and Trepte, C.R., 1995, Atmospheric effects of the Mt. Pinatubo eruption: Nature, v. 373, p. 399-404.
McPeters, R. D., 1993, The atmospheric SO2 budget for Pinatubo derived from NOAA-11 SBUV/2 spectral data: Geophysical Research Letters, v. 20, p. 1971-1974.
Metrich, N., and Rutherford, M. J., 1992, Experimental study of chlorine behavior in hydrous silicic melts: Geochimica et Cosmochimica Acta, v. 56, p. 607-616.
Mori, J., Eberhart-Phillips, D., and Harlow, D., this volume, Three-dimensional velocity structure at Mount Pinatubo, Philippines: Resolving magma bodies and earthquake hypocenters.
Newman, S., Epstein, S., and Stolper, E., 1988, Water, carbon dioxide, and hydrogen isotopes in glasses from the ca. 1340 A.D. eruption of the Mono Craters, California: Constraints on degassing phenomena and initial volatile content: Journal of Volcanology and Geothermal Research, v. 35, p. 75-96.
Palais, J. M., and Sigurdsson, H., 1989, Petrologic evidence of volatile emissions from major historic and pre-historic volcanic eruptions, in Kidson, J. W. ed., Understanding climate change: American Geophysical Union Monograph 52, p. 31-53.
Pallister, J. S., Hoblitt, R. P., and Reyes, A. G., 1992, A basalt trigger for the 1981 eruptions of Pinatubo volcano?: Nature, v. 356, p. 426-428.
Pallister, J. S., Hoblitt, J. S., Meeker, G. P., Newhall, C. G., Knight, R. J., and Siems, D. F., this volume, Magma mixing at Mount Pinatubo volcano: Petrographic and chemical evidence from the 1991 deposits.
Pan, V., Holloway, J. R., and Hervig, R. L., 1991, The temperature and pressure dependence of carbon dioxide solubility in tholeiitic basalt: Geochimica et Cosmochimica Acta, v. 55, p. 1587-1595.
Pasteris, J. D., Wopenka, B., Wang, A., and Harris, T. N., this volume, Relative timing of fluid and anhydrite saturation: Another consideration in the sulfur budget of the Mount Pinatubo eruption.
Prather, M., 1992, Catastrophic loss of stratospheric ozone in dense volcanic clouds: Journal of Geophysical Research, v. 97, p. 10,187-10,191.
Qin, Z., Lu, F., and Anderson, A. T., 1992, Diffusive reequilibration of melt and fluid inclusions: American Mineralogist, v. 77, p. 565-576.
Read, W. G., Froidevaux, L., and Waters, J. W., 1993, Microwave limb sounder measurement of stratospheric SO2 from the Mt. Pinatubo volcano: Geophysical Research Letters, v. 20, p. 1299-1302.
Rose, W. I., 1977, Scavenging of volcanic aerosol by ash: Atmospheric and volcanic implications: Geology, v. 5, p. 621-624.
Ruaya, J. R., Ramos, M. N., and Gonfiantini, R., 1992, Assessment of magmatic components of the fluids at Mt. Pinatubo volcanic-geothermal system, Philippines from chemical and isotopic data: Geological Survey of Japan Report, no. 279, p. 141-151.
Rutherford, M. J., and Devine, J. D., 1991, Preeruption conditions and volatiles in the 1991 Pinatubo magma [abs.]: Eos, Transactions, American Geophysical Union, v. 72, p. 62.
Rutherford, M. J., and Devine, J. D., this volume, Preeruption P-T conditions and volatiles in the 1991 Mount Pinatubo magma.
Scott, W. E., Hoblitt, R. P., Torres, R., Martinez, M., Nillos, T., and Self, S., this volume, Pyroclastic flows of the June 15, 1991, paroxysmal eruption of Mount Pinatubo volcano, Philippines.
Shinohara, H., Iiyama, J. T., and Matsuo, S., 1989, Partition of chlorine compounds between silicate melt and hydrothermal solutions: I. Partition of NaCl-KCl: Geochimica et Cosmochimica Acta, v. 53, p. 2617-2630.
Sigurdsson, H., 1990, Assessment of the atmospheric impact of volcanic eruptions, in Sharpton, V. L., and Ward, P. D., eds., Global catastrophies in Earth history: Geological Society of America Special Paper 247, p. 99-110.
Sigurdsson, H., Carey, S., Palais J. M., and Devine, J. D., 1990, Preeruption composition gradients and mixing of andesite and dacite magma erupted from Nevado del Ruiz volcano, Colombia in 1985: Journal of Volcanology and Geothermal Research, v. 41, p. 127-151.
Sigurdsson, H., Devine, J. D., and Davis, A. N., 1985, The petrologic estimation of volcanic degassing: Jokull, v. 35, p. 1-8.
Silver, L. A., Ihinger, P. D., and Stolper, E., 1990, The influence of bulk composition on the speciation of water in silicate glasses: Contributions to Mineralogy and Petrology, v. 104, p. 142-162.
Skirius, C. M., Peterson, J. W, and Anderson, A. T., 1990, Homogenizing rhyolitic glass inclusions from the Bishop Tuff: American Mineralogist, v. 75, p. 1381-1398.
Solomon, S., Sanders, R. W., Garcia, R. R., and Keys, J. G., 1993, Increased chlorine dioxide over Antarctica caused by volcanic aerosols from Mount Pinatubo: Nature, v. 363, p. 245-248.
Sparks, R. S. J., Barclay, J., Jaupart, C., Mader, L., and Phillips, J.C., 1994, Physical aspects of magmatic degassing I. Experimental and theoretical constraints on vesiculation, in Carroll, M. R., and Holloway, J. R., eds., Volatiles in magmas: Mineralogical Society of America Reviews in Mineralogy, v. 30, p. 413-445.
Stoiber, R. E., and Jepsen, A., 1973, Sulfur dioxide contributions to the atmosphere by volcanoes: Science, v. 182, p. 577-578.
Swanson, D. A., and Holcomb, R. T., 1989, Regularities in growth of the Mount St. Helens dacite dome, 1980-1986: IAVCEI Proceedings in Volcanology, v. 2, p. 1-24.
Symonds, R. B., and Reed, M. H., 1993, Calculation of multicomponent chemical equilibria in gas-solid-liquid systems: Calculation methods, thermochemical data, and applications to studies of high-temperature volcanic gases with examples from Mount St. Helens: American Journal of Science, v. 293, p. 758-864.
Symonds, R. B., Rose, W. I., Bluth, G., J. S., Gerlach, T. M., 1994, Volcanic-Gas studies: methods, results, and applications, in Carroll, M. R., and Holloway, J. R., eds., Volatiles in Magmas: Mineralogical Society of America Reviews in Mineralogy, v. 30, p. 1-66.
Tait, S., 1992, Selective preservation of melt inclusions in igneous phenocrysts: American Mineralogist, v. 77, p. 146-155.
Varekamp, J. C., Luhr, J. F., and Prestegaard, K. L., 1984, The 1982 eruptions of El Chichón volcano (Chiapas, Mexico): Character of the eruptions, ash-fall deposits, and gas phase: Journal of Volcanology and Geothermal Research, v. 23, p. 39-68.
Wallace, P., and Anderson, A. T., 1994, Preeruptive gradients in H2O, CO2, and exsolved gas in the magma body of the Bishop Tuff [abs.]: Eos, Transactions, American Geophysical Union, v. 75, no. 44, p. 719.
Wallace, P., and Gerlach, T., M., 1994, Magmatic vapor source for sulfur dioxide released during volcanic eruptions: Evidence from Mount Pinatubo: Science, v. 265, p. 497-499.
Webster, J. D., 1992a, Fluid-melt interactions involving Cl-rich granites: Experimental study from 2 to 8 kbar: Geochimica et Cosmochimica Acta, v. 56, p. 659-678.
Webster, J. D., 1992b, Water solubility and chlorine partitioning in Cl-rich granitic systems: Effects of melt composition at 2 kbar and 800°C: Geochimica et Cosmochimica Acta, v. 56, p. 679-687.
Webster, J. D., and Holloway, J. R., 1990, Partitioning of F and Cl between hydrothermal fluids and highly evolved granitic magmas, in Stein, H. J., and Hannah, J. L., eds., Ore-bearing granite systems: Petrogenesis and mineralizing processes: Geological Society of America Special Paper 246, p. 21-34.
Westrich, H. R., and Gerlach, T. M., 1992, Magmatic gas source for the stratospheric SO2 cloud from the June 15, 1991 eruption of Mount Pinatubo: Geology, v. 20, p. 867-870.
Williams, S. N., Sturchio, N. C., Calvache V., M. L., Mendez F., R., Londono C., A., and Garcia P., N., 1990, Sulfur dioxide from Nevado del Ruiz volcano, Colombia: Total flux and isotopic constraints on its origin: Journal of Volcanology and Geothermal Research, v. 42, p. 53-68.
Williams, S. N., Schaefer, S. J., Calvache, M. L. and Lopez, D., 1992, Global carbon dioxide emission to the atmosphere by volcanoes: Geochimica et Cosmochimica Acta, v. 56, p. 1765-1770.
Wilson, L., Sparks, R. S. J., and Walker, G. P. L., 1980, Explosive volcanic eruptions--IV. The control of magma properties and conduit geometry on eruption column behaviour: Geophysical Journal Royal Astronomical Society, v. 63, p. 117-148.
Wolfe, E. W., and Hoblitt, R. P., and others, this volume, Overview of the eruptions.
FIRE and MUD Contents
PHIVOLCS | University of Washington Press | U.S.Geological Survey
This page is <https://pubs.usgs.gov/pinatubo/gerlach/>
Contact: Chris Newhall
Last updated 06.10.99