Many natural factors can affect ground-water quality; however, the primary factors include the source and chemical composition of recharge water, the lithological and hydrological properties of the geologic unit, the various chemical processes occurring within the geologic unit, and the amount of time the water has remained in contact with the geologic unit (residence time). All of these factors can affect the type and quantities of dissolved constituents in ground water. The most abundant dissolved constituents measured are the major ions, which can be both positively charged (cations) and negatively charged (anions). Because of the requirements of electroneutrality, cations and anions are present at equal concentrations in water and comprise most of the dissolved solids in ground water. The most abundant cations present in water are calcium (Ca), magnesium (Mg), sodium (Na), and potassium (K); the most abundant anions are bicarbonate (HCO3), chloride (Cl), and sulfate (SO4). By measuring the concentrations of these ions in ground-water samples, the ionic composition of the water is determined and the chemical quality of the water can be characterized and described. A brief summary of the source or cause of these and other dissolved constituents and physical and general mineral characteristics commonly present in ground water is presented in table 3.
The ionic composition of water is used to classify it into ionic types based on the dominant dissolved cation and anion, expressed in milliequivalents per liter (meq/L). A milliequivalent (meq) is a measurement of the molar concentration of the ion, normalized by the ionic charge of the ion. The dominant dissolved ion must be greater than 50 percent of the total. For example, water classified as a sodium-bicarbonate-type water contains more than 50 percent of the total cation milliequivalents as sodium and more than 50 percent of the total anion milliequivalents as bicarbonate. If no cation or anion is dominant (greater than 50 percent), the water is classified as mixed and the two most common cations or anions in decreasing order of abundance are used to describe the water type. For example, a water containing 45 percent sodium, 35 percent calcium, and 20 percent magnesium, and 55 percent bicarbonate, 30 percent sulfate, and 15 percent chloride would be classified as a sodium-calcium-bicarbonate-type water.
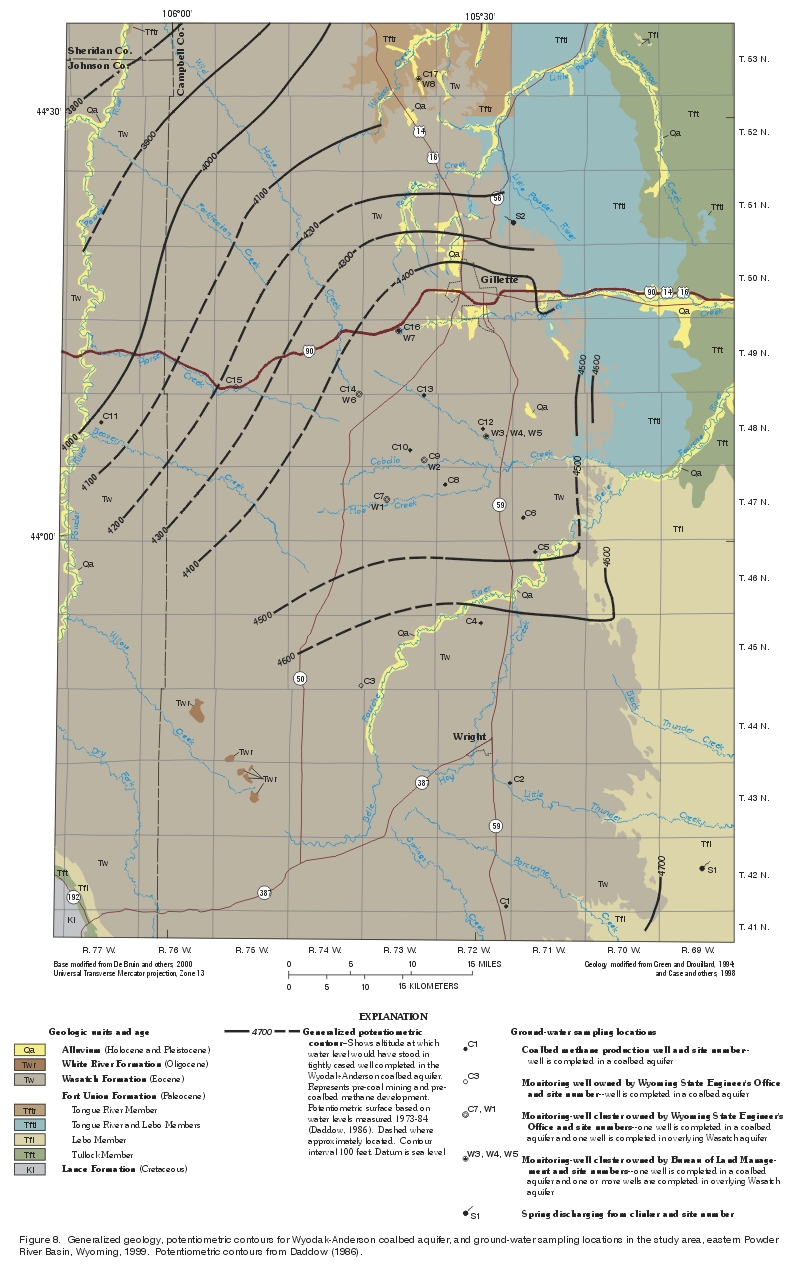
A review of water-quality analyses (appendix table 1) in this study suggested that four sampled monitoring wells (C3, C7, C9, and C14, fig. 8) were contaminated by grout during well completion. The pH values for ground-water samples collected from these wells were very alkaline (pH = 9.6, 10.5, 11.3, and 11.7), were much higher than for similar wells completed in the coalbed aquifers in the study area (Rice and others, 2000), and were uncommonly high for natural aquifers (Hem, 1985). High pH values in this range are typical for wells contaminated with alkaline grout (cement and/or bentonite) used to seal the annular space during well completion (Nielson, 1991). Nielson (1991, p. 585) described four ways that very alkaline water from annular seals can enter well screen areas and be sampled: "(1) Wells located in low permeability units with strong vertical gradients (i.e., grout bleeds directly downward); (2) Grout injected into the screened area of the well; (3) Bentonite seals too thin or ineffective; and (4) Fractured rock providing channels around bentonite seals." It is unclear which of these may have affected the water-quality analyses, but the approach used in this investigation was to remove all water-quality analyses for samples from these four wells.
Ion balances were calculated and examined for each ground-water sample as a quality-assurance check of the chemical analyses. The ion balance was calculated (in meq/L) as the total dissolved-cation concentration minus the total dissolved-anion concentration divided by the total concentration of ions dissolved in solution. The total cation concentration was calculated as the sum of calcium, magnesium, sodium, and potassium; the total anion concentration was calculated as the sum of acid-neutralizing capacity, chloride, fluoride, and sulfate. Inorganic nitrogen species (nitrite plus nitrate and ammonium) were not included in the calculations because neither was measured in the ground-water samples. Nineteen of twenty-one ground-water samples (about 90 percent) had ion balances within the +/- 6 percent range, indicating that the major-ion analyses were of good quality. The remaining two samples had ion balances of about 9 and 12 percent; these samples are still included and considered acceptable in this study because the ion balances are still relatively low, and unmeasured constituents such as organic anions, nutrients, and trace metals may contribute to higher ion balances (Hem, 1985).
A replicate sample (duplicate) is a ground-water quality-control sample collected sequentially after a regular sample (normal environmental sample); both are analyzed for the same constituents to assess the combined effects of field and laboratory procedures on measurement variability. Constituents measured in both samples are compared by calculating the relative-percent difference (RPD) using the equation given below. The RPD cannot be calculated for compounds that are below the minimum reporting level (MRL).

Two replicate samples were collected during this study. Results of the RPD calculations are shown in table 4. RPD values greater than zero but less than 1 percent are reported as less than 1 percent (<1 percent) in the table.
For the first replicate sample, RPDs for all but two measured constituents were less than 1 percent in one normal environmental sample and associated replicate sample, indicating very good precision (reproducibility) (table 4). The RPD was 4 percent for calcium and 8 percent for potassium. In the second sample, RPDs for all but five constituents were less than 1 percent. RPDs for magnesium, sodium, chloride, and silica were about 3 to 6 percent. The RPD for tritium is high (about 40 percent), indicating poor precision between the two sample types; the source of the poor precision is unclear. Therefore, precision between all constituents but tritium is very good in the second set of samples.
Ground-water-quality data for samples collected from springs and wells are presented in the Appendix, and summary statistics for samples collected from wells are presented in tables 5 and 6. Visual descriptions and comparisons of data collected from springs and wells in the Wasatch aquifer are provided by plotting individual data values because the sample sizes are less than 10 (figs. 10, 11, and 12). Percentiles and boxplots were constructed for characteristics and constituents measured, analyzed, or calculated in samples collected from coalbed aquifers.

Figure 10. Physical characteristics of ground water, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 99 kb) |

Figure 11. General mineral characteristics of ground water, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 199 kb) |

Figure 12. Concentrations of major ions in ground water, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 205 kb) |
Figure 12. Continued. (Click on image for a larger version, 156 kb) |
Dissolved solids are reported as a concentration value in mg/L (milligrams per liter) calculated by summing all major ions; dissolved-solids concentrations often are used to compare water quality between different groups of water samples. Dissolved-solids concentrations were used to compare data collected as part of this study to data collected by several other investigations with larger sample sizes (n>30).
Despite small sample size (n = 7), the median dissolved-solids concentration for ground-water samples collected from wells completed in the Wasatch aquifer in the study area (1,010 mg/L) is the same (1,010 mg/L) as reported by Lowry, Wilson, and others (1986) for the Powder River drainage basin in Wyoming and Montana, and similar to the median dissolved-solids concentration (1,220 mg/L) reported by Larson (1984) for samples collected from the Wasatch aquifer in Campbell County (Wyoming), suggesting good comparability. In contrast, the median dissolved-solids concentration (1,010 mg/L) is about one-half the median dissolved-solids concentration (2,215 mg/L) reported by Martin and others (1988) for samples collected from wells completed in the Wasatch Formation in the Powder River Basin. The difference may be attributable to the location and depth of the wells examined by Martin and others (1988). Those wells were located at coal mines at relatively shallow depths along the eastern margin of the study area. As will be discussed later in this report, the ionic composition of the shallow part of the Wasatch Formation is believed to be different than deeper parts of the formation; this apparent relationship between depth and ionic composition in the Wasatch Formation may explain the observed differences in median dissolved-solids concentrations between this study and the study by Martin and others (1988).
The median dissolved-solids concentration for 13 ground-water samples collected from wells completed in the coalbed aquifers (644 mg/L) is lower than reported by Martin and others (1988), 1,310 mg/L for 379 samples collected from 88 wells at coal mines. The 88 wells were located at the coal mines at relatively shallow depths along the eastern margin of the study area. Water quality of the coalbed aquifers in this area may be different because of the location of the wells in relation to clinker deposits and/or the burn line (the contact between the unburned coal or overburden) (Heffern and Coates, 1999); this will be discussed later in the report. In contrast, the median dissolved-solids concentration (644 mg/L) in this study is very similar to the median dissolved-solids concentration calculated for ground-water samples (680 mg/L) collected from 36 wells completed in coalbed aquifers in the study area as part of another investigation (Rice and others, 2000). That dataset represents a better comparison because ground-water samples were collected from coalbed aquifers throughout the study area.
|
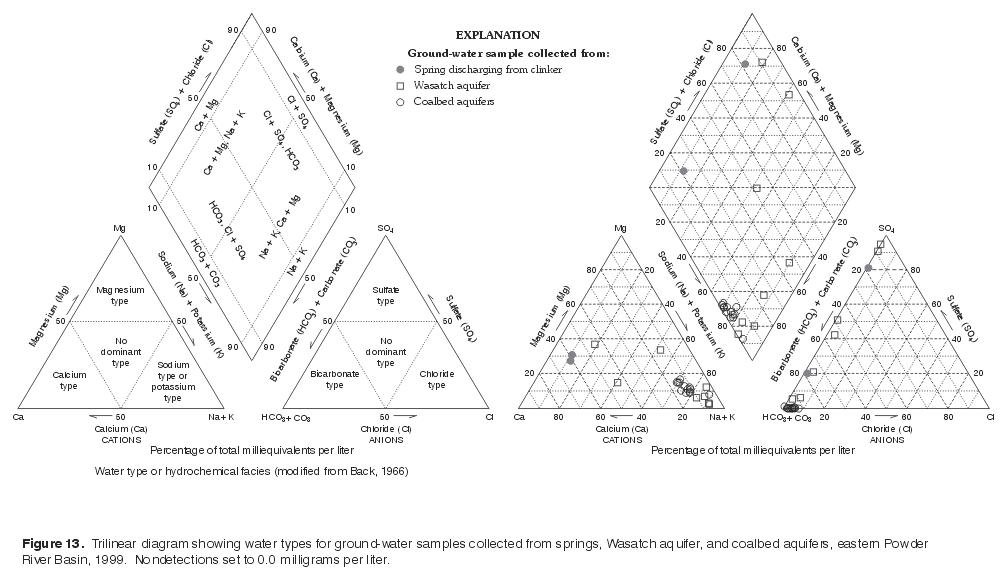
The relative ionic composition of ground-water samples collected from springs
and wells in the study area are plotted on a trilinear diagram (fig. 13). A
trilinear diagram, also frequently referred to as a Piper diagram (Piper, 1944),
provides a convenient method to classify and compare water types based on the
ionic composition of different water samples (Hem, 1985). Cation and anion
concentrations for each ground-water sample are converted to total meq/L and
plotted as percentages of their respective totals in two triangles
(fig. 13). The cation and anion relative percentages in each triangle are
then projected into a quadrilateral polygon that describes the water type or
hydrochemical facies.
Samples collected from two springs in the study area both had the same dominant cation, calcium, but different dominant anions, sulfate and bicarbonate (fig. 13); samples collected from these springs would be classified as calcium-sulfate-type and calcium-bicarbonate-type waters. |
Figure 13. Trilinear diagram showing water types for ground-water samples collected from springs, Wasatch aquifer, and coalbed aquifers, eastern Powder River Basin, 1999. Nondetections set to 0.0 milligrams per liter. (Click on image for a larger version, 205 kb) |
Ground-water samples collected from wells had a different ionic composition than samples collected from the springs, but had very similar compositions with respect to each other, regardless of aquifer (fig. 13). All ground-water samples collected from the coalbed aquifers were sodium-bicarbonate-type waters and only three of eight samples collected from the Wasatch aquifer were not classified the same. Coalbed aquifers in the Powder River Basin in Wyoming and Montana are characterized by sodium-bicarbonate-type waters, as noted by other investigators (Law and others, 1991; VanVoast, 1991; Rice and others, 2000; Rice, 2000). Two of the three samples collected from the overlying Wasatch aquifer that were not sodium-bicarbonate-type waters were similar in ionic composition, with mixed cations, and with the same dominant anion, sulfate. The other sample was a mixed type, and plots in the sodium-magnesium-sulfate-bicarbonate part of the trilinear diagram. The similarity in water type between most ground-water samples suggests that similar geochemical processes may be controlling major-ion chemistry in these aquifers and that the waters had the same or similar origins.
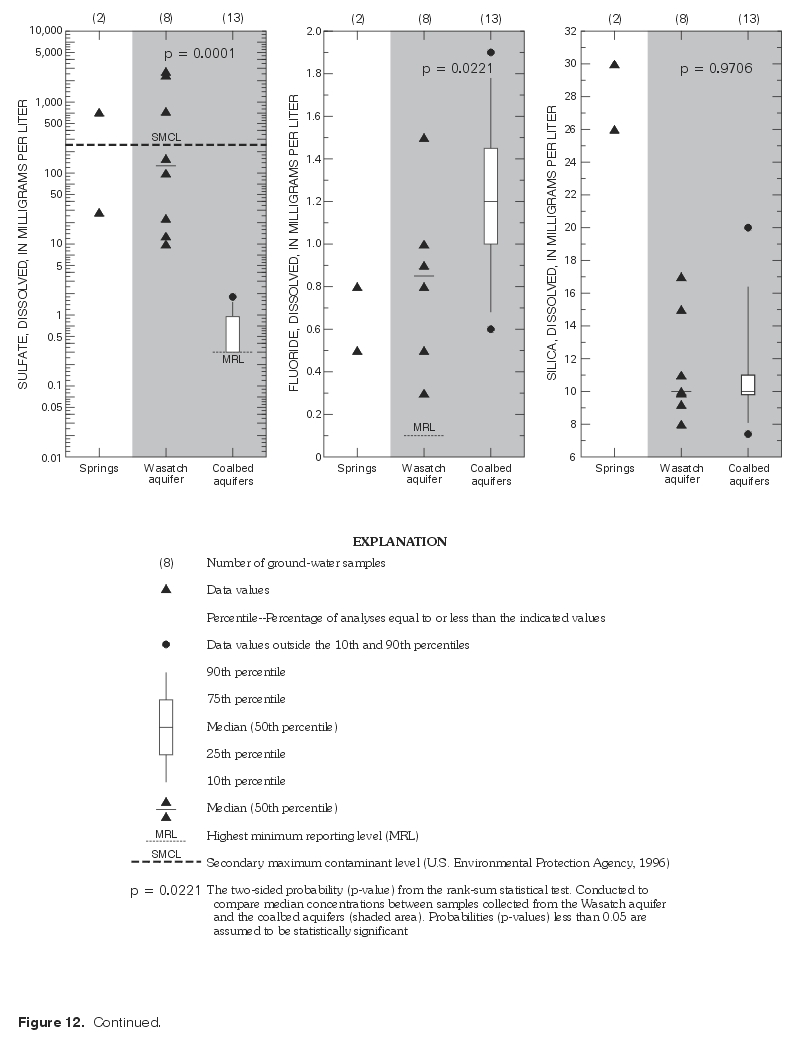
The two-sided version of the rank-sum test (Helsel and Hirsch, 1992), also known as the Wilcoxon rank-sum test (Wilcoxon, 1945) or Mann-Whitney test (Mann, 1945), is a nonparametric hypothesis test that was used to determine the probability that the median dissolved-solids and major-ion concentrations between aquifers were the same. The rank-sum test, like most nonparametric hypothesis tests, requires no assumptions about the population distribution, is resistant to outliers, and is more appropriate for small sample sizes where nonnormality is more difficult to detect. The null hypothesis of identical median concentrations between aquifers was rejected if the probability (p-value) of obtaining identical medians by chance was less than 0.05. Results of the hypothesis testing are provided in table 7 and in figure 12.
Median concentrations of all but two major ions were statistically the same between coalbed and overlying aquifers (p-values > 0.05). Fluoride concentrations were significantly different between aquifers (p-value = 0.0221). It is unclear why fluoride concentrations differ between aquifers, although it has been suggested that differences in fluoride concentrations within the Wasatch and Fort Union Formations may be due to local lithological variation (Wyoming Water Development Commission, 1985). While median sulfate concentrations are low in the overlying Wasatch aquifer (130 mg/L), sulfate concentrations are very low in samples collected from the coalbed aquifers, as indicated by greater than 50 percent of data values at concentrations less than detectable limits. This contrast is easily seen on the boxplot and plotting of individual data values presented earlier (fig. 12). Results of the rank-sum test indicate that these differences are statistically significant (p-value = 0.0001) between aquifers. It should be noted that the severe censoring of sulfate concentrations in samples collected from the coalbed aquifers may reduce the ability of the rank-sum test to reject the null hypothesis when actually false (Helsel and Hirsch, 1992); however, the very low p-value (0.0001) for sulfate would indicate that this is not the case.
|
The ionic composition of ground-water samples can be represented by another
type of water-quality diagram - the stiff diagram (Stiff, 1951). Stiff
diagrams are used to compare the ionic composition of water samples between
different locations, depths, or aquifers. The stiff diagram is a polygon created
from three horizontal axes extended on both sides of a vertical axis. Cations
are plotted on the left side of the axis and anions are plotted on the right
side, both in meq/L. A greater distance from the vertical axis represents a
larger ionic concentration. The cation and anion concentrations are connected
to form an asymmetric polygon known as a stiff diagram, where the size is a
relative indication of the dissolved-solids concentration.
The areal distribution of stiff diagrams constructed for all water samples collected as part of this study are shown in figure 14. The horizontal axis units are the same on all stiff diagrams to aid comparisons between different sampling locations and aquifers. The color of the stiff diagrams indicates the aquifer and the number above each diagram is the calculated dissolved-solids concentration. Like the trilinear diagram presented and discussed earlier, the stiff diagrams show and reinforce the similarity in ionic composition (sodium-bicarbonate-type water) observed in most ground-water samples, regardless of aquifer. Stiff diagrams constructed for samples collected from wells completed in the Wasatch aquifer show no areal pattern in water type (all but three are sodium-bicarbonate-type waters) or obvious areal pattern in dissolved-solids concentrations; this suggests that well location, in addition to aquifer, may not be related to water type. However, there may be a relationship between well depth and ionic composition in the Wasatch aquifer. |
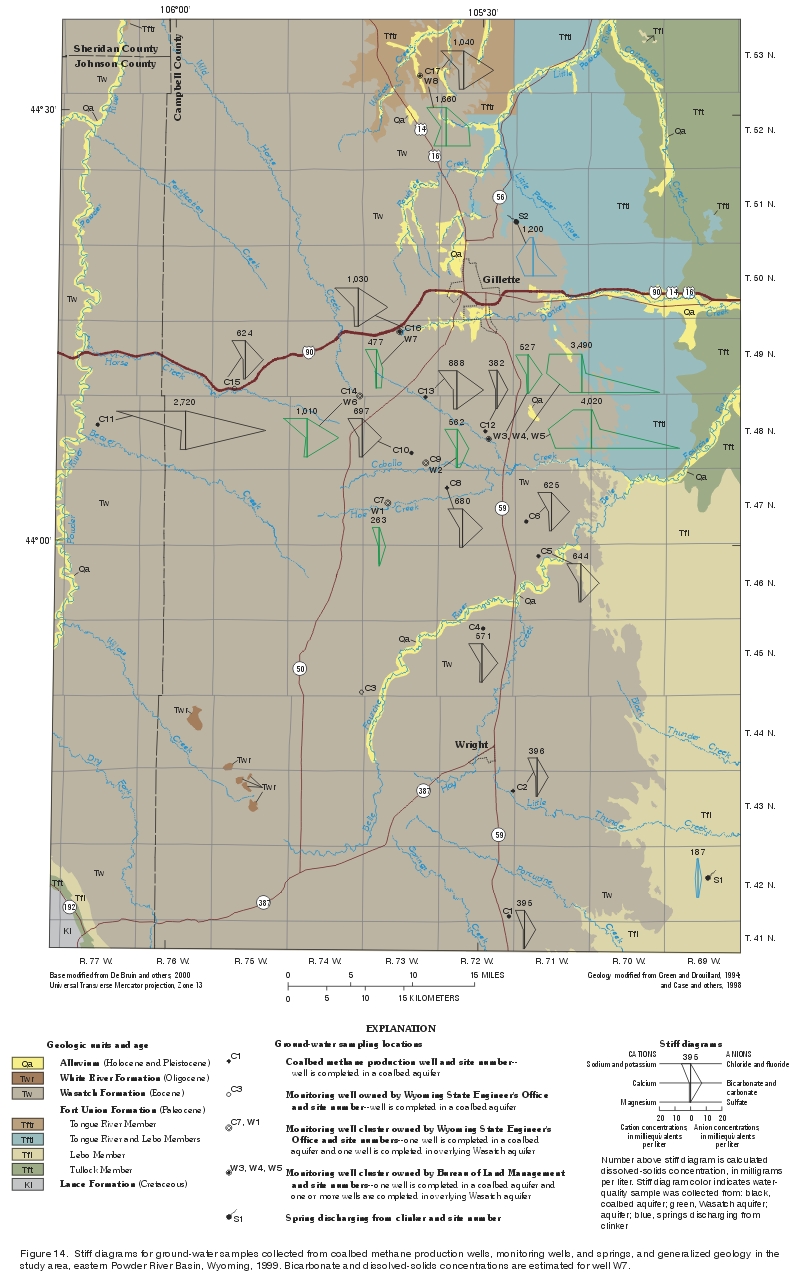
Figure 14. Stiff diagrams for ground-water samples collected from coalbed methane production wells, monitoring wells, and springs, and generalized geology in the study area, eastern Powder River Basin, Wyoming, 1999. Bicarbonate and dissolved-solids concentrations are estimated for well W7. (Click on image for a larger version, 442 kb) |
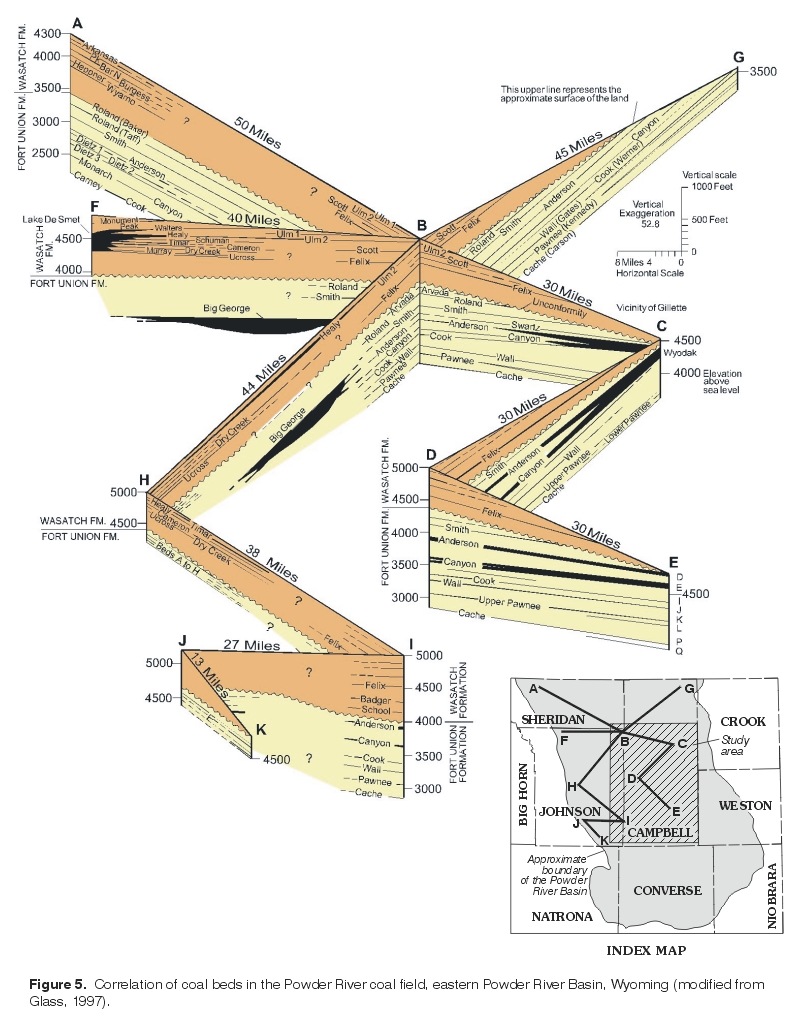
Stiff diagrams constructed for ground-water samples collected from wells completed in the coalbed aquifers also show no spatial pattern because the water type is the same (sodium-bicarbonate type) for all samples. The dissolved-solids concentration in the sample collected from well C11 (fig. 14), the most western site in the study area, is much higher than all other samples collected from wells completed in coalbed aquifers; this sample was collected from a well completed in the Big George coal bed, a coal bed considered equivalent to the Wyodak-Anderson coal zone (Flores and Bader, 1999). The Big George coal bed is both deeper and possibly older (Flores, 1999) than other coal beds in the Wyodak-Anderson coal zone (fig. 5). It should be noted that this well also is completed in sandstone and shale overlying the coal bed, and the effect on resulting water quality cannot be determined. However, another water-quality sample collected from this coal bed in the same area was similar in both water type (sodium-bicarbonate type) and high dissolved-solids concentration (Rice and others, 2000).
On the basis of ground-water samples collected during this study and additional samples collected in the area as part of another study (Rice and others, 2000), dissolved-solids concentrations in the Wyodak-Anderson coalbed aquifer appear to be lower (generally less than 600 mg/L) south of the Belle Fourche River, near the southern Rochelle Hills (an area with clinker in the Lebo Member of the Fort Union Formation). Clinker in this area has been speculated to provide much recharge to coalbed aquifers downdip to the west in the study area (Davis, 1976; Daddow, 1986; Lowry, Wilson, and others, 1986; Martin and others, 1988; Heffern and others, 1996; Bureau of Land Management, 1999; Heffern and Coates, 1999).
Dissolved-solids concentrations represent total ions dissolved in solution; concentrations typically are lower near recharge sources where ground-water residence times (time in contact with aquifer materials) are relatively short and increase as ground water flows away from the source of recharge and residence times increase. As stated earlier, potentiometric contours suggest regional ground-water flow in the Wyodak-Anderson coalbed aquifer may be towards the west and northwest, away from clinker in the area (Daddow, 1986) (fig. 8). If the clinker in this area is the source of most of the recharge to the coalbed aquifers in the study area, dissolved-solids concentrations should increase as ground water flows away from the clinker deposits, a pattern that may be present and may explain the apparent increase in dissolved-solids concentrations as ground water flows north of the Belle Fourche River. Alternatively, the lower dissolved-solids concentrations in the area may not represent a true spatial pattern and may simply be the result of natural or random variation, different processes in the area, or a product of small sample size in the area.
Dissolved-solids concentrations in the Wyodak-Anderson coalbed aquifer appears to increase northeast of the Belle Fourche River and outside of the study area. Rice and others (2000) noted that dissolved-solids concentrations in the Wyodak-Anderson coalbed aquifer appears to increase from south to north and from east to west in the Powder River Basin in Wyoming. The investigators suggest that "this trend may be a result of increased water-rock interaction along a flowpath, an increase or change in composition of the ash content of the coal, or other factors not yet recognized" and "the increase in TDS (total dissolved solids) is generally a result of an increase in sodium and bicarbonate content of the water" (Rice and others, 2000, p. 4).
|
Dissolved-solids and major-ion concentrations in ground-water samples collected from wells were compared with well depth to determine if there was a statistically significant relationship. Kendall's tau test statistic (Helsel and Hirsch, 1992), a nonparametric measure of the monotonic correlation between two continuous variables, was calculated to measure the strength of this relationship. Kendall's tau is a correlation coefficient calculated using the ranks of the data rather than actual data values; therefore, the test statistic is resistant to outliers and can be used for datasets with moderate censoring. Tau is dimensionless and ranges between -1 and 1; when one variable increases as the second increases, tau is positive. If the variables vary in opposite directions, tau is negative. If a strong correlation is observed, tau, like any statistical correlation coefficient, does not explain the cause of the relationship. Kendall's tau correlation coefficients were considered statistically significant when probabilities (p-values) were less than 0.05; the null hypothesis of no correlation between dissolved-solids or major-ion concentrations and well depth was rejected if the probability of obtaining this correlation by chance was less than this value. The exact form of the test was used for the samples collected from the Wasatch aquifer because the sample size was less than 10; the large sample approximation was used for samples collected from the coalbed aquifers because sample size was greater than 10. Kendall's tau correlation coefficients and resulting probabilities (p-values or attained levels of significance) between dissolved solids or major ions and well depth for eight samples collected from the Wasatch aquifer are shown in table 8. Bicarbonate and fluoride were positively correlated with well depth; all other major ions were negatively correlated with well depth. Correlation between calcium and well depth is very strong (tau = -0.93) and statistically significant (p-value = 0.0004); correlation between sulfate and well depth was strong (tau = -0.79) and also is statistically significant (p-value = 0.0056). These statistically significant correlations between calcium and sulfate and well depth are illustrated in scatterplots (figure 15). The correlation between dissolved solids and well depth is relatively strong (tau = -0.62) and close to statistical significance (p-value = 0.07). Similar nonsignificant correlations were observed between magnesium and well depth (tau = -0.57, p-value = 0.062) and fluoride and well depth (tau = 0.54, p-value = 0.084).
|

Figure 15. Calcium and sulfate concentrations in relation to well depth, Wasatch aquifer, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 187 kb)
|
| Kendall's tau correlation coefficients and resulting probabilities
(p-values) between dissolved solids or major ions and well depth for 13 samples
collected from coalbed aquifers are shown in table 9. Chloride and fluoride were
negatively correlated with well depth; all other major ions were positively
correlated with well.
The strongest correlation was observed between potassium and well depth; this positive correlation (tau = 0.462) is statistically significant (p-value = 0.0256), but not strong. The statistically significant correlation between potassium and well depth is illustrated in a scatterplot (fig. 16). Kendall's tau was not calculated for sulfate because greater than 50 percent of the data values were censored (less than the MRL). |
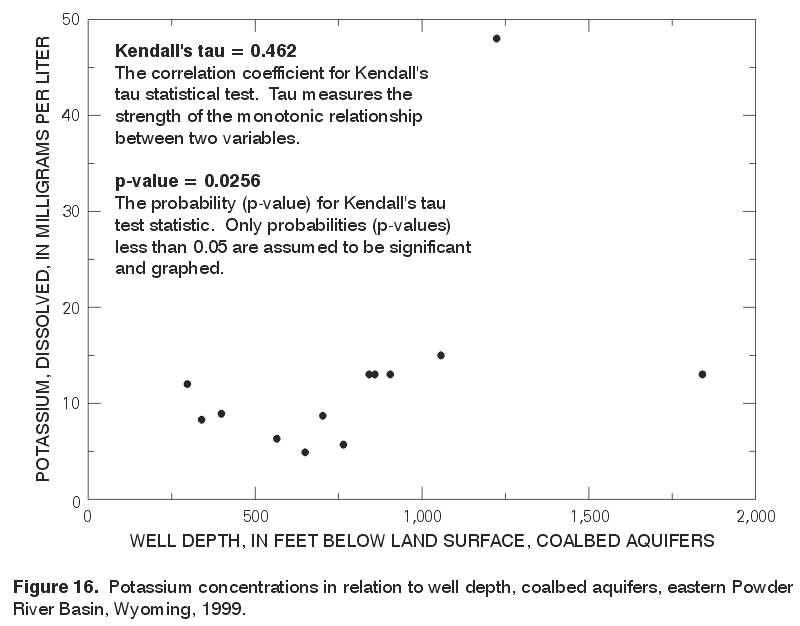
Figure 16. Potassium concentrations in relation to well depth, coalbed aquifers, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 108 kb) |
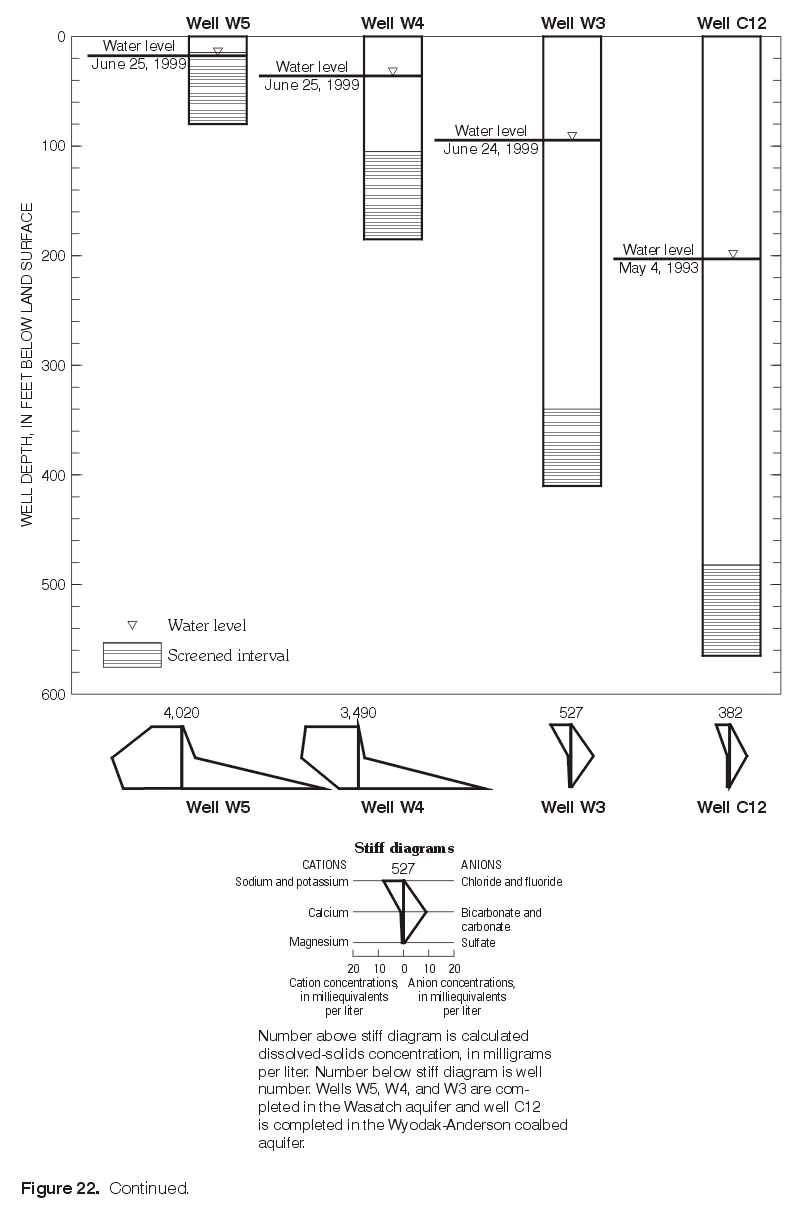
Major-ion chemistry in the vertical dimension was examined qualitatively in three areas with monitoring-well clusters, including one area with a monitoring-well cluster where three wells are completed in the Wasatch aquifer (W5, W4, W3) and overlie a nearby well (C12) completed in the Wyodak-Anderson coalbed aquifer (fig. 14); each well is completed progressively deeper (fig. 17). Water-level measurements indicated the hydraulic potential for downward vertical ground-water flow at this location. Major-ion composition was similar in ground-water samples collected from the two shallowest wells (W5 and W4) with both having mixed cations (calcium, magnesium, and sodium) and the same dominant anion, sulfate. The ionic composition was similar in samples collected from the two deepest wells (W3 and C12)—both were sodium-bicarbonate-type waters. As illustrated in figure 17, calcium, magnesium, and sulfate concentrations decrease with increasing well depth; these decreases correspond with a large decrease (from 4,020 mg/L to 382 mg/L) in dissolved-solids concentrations with increasing depth. Sodium concentrations increase with depth between the two shallowest wells (W5 and W4) but decrease with increasing well depth in all the other wells (W4, W3, and C12) completed successively deeper. Bicarbonate concentrations decrease with increasing well depth in the two shallowest wells (W5 and W4), increase with depth between the next two deepest wells (W4 and W3), and then decrease between the two deepest wells (W3 and C12).
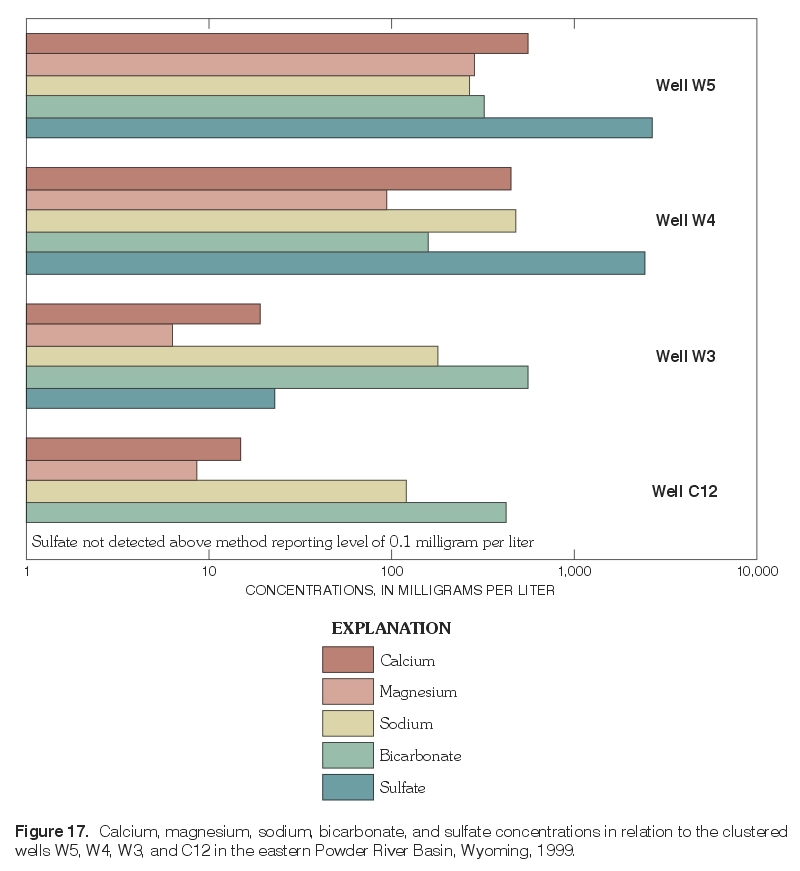
Figure 17. Calcium, magnesium, sodium, bicarbonate, and sulfate concentrations in relation to the clustered wells W5, W4, W3, and C12 in the eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 107 kb) |
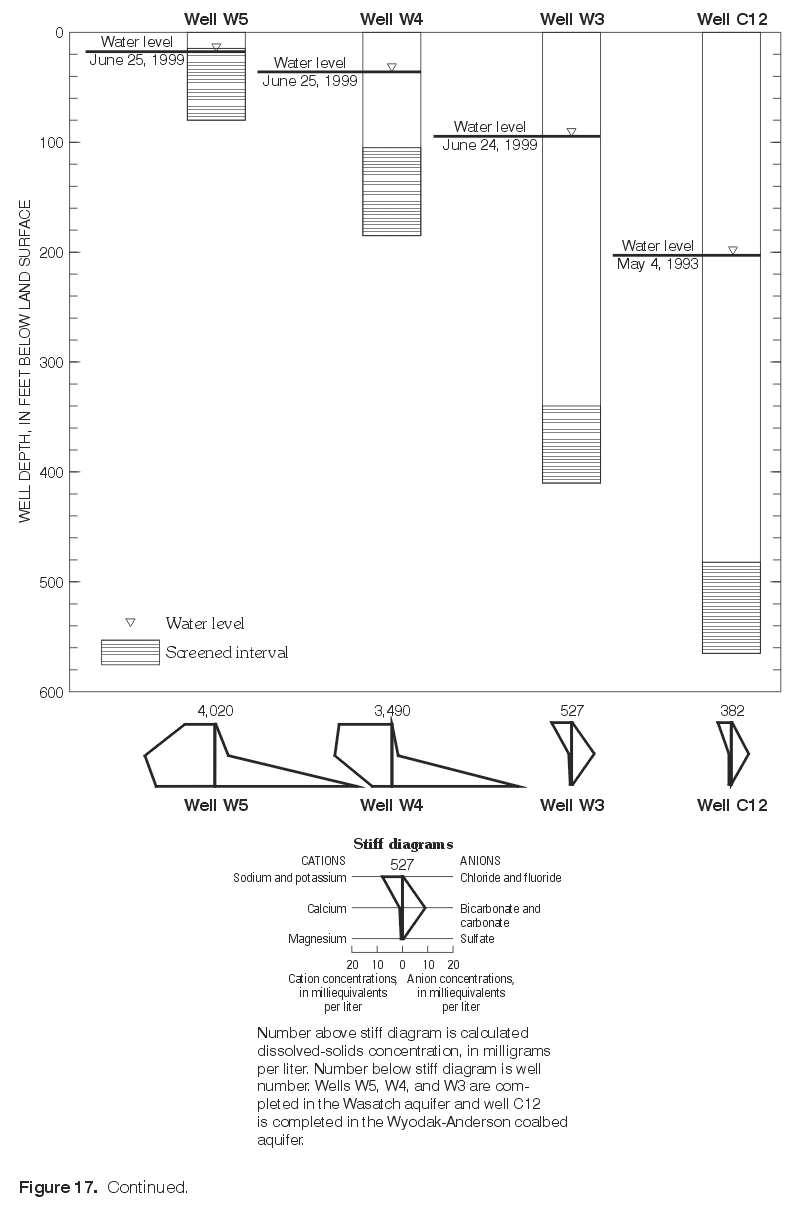
Figure 17. Continued. (Click on image for a larger version, 142 kb) |

Figure 18. Calcium, magnesium, sodium, bicarbonate, and sulfate concentrations in relation to the clustered wells W8 and C17 in the eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 77 kb) |
Figure 18. Continued. (Click on image for a larger version, 123 kb) |
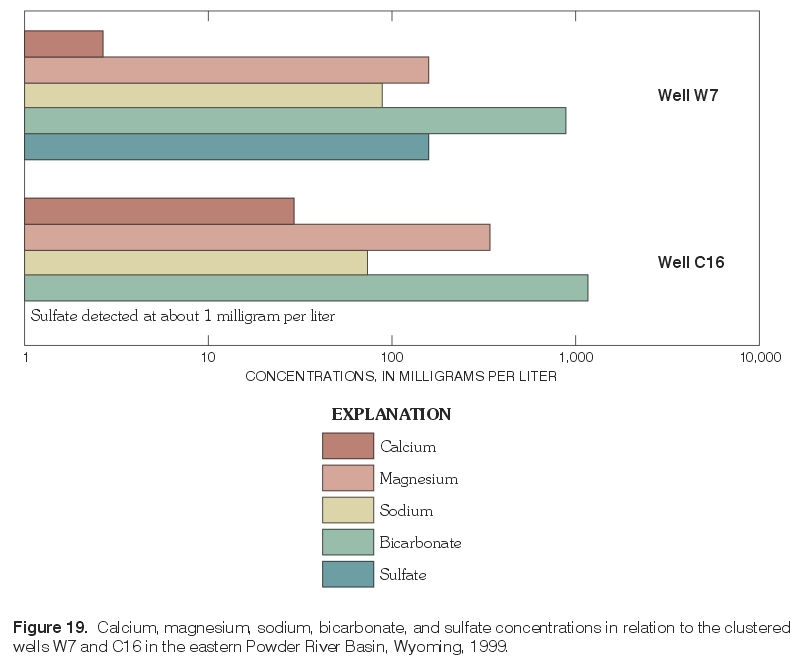
Figure 19. Calcium, magnesium, sodium, bicarbonate, and sulfate concentrations in relation to the clustered wells W7 and C16 in the eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 77 kb) |
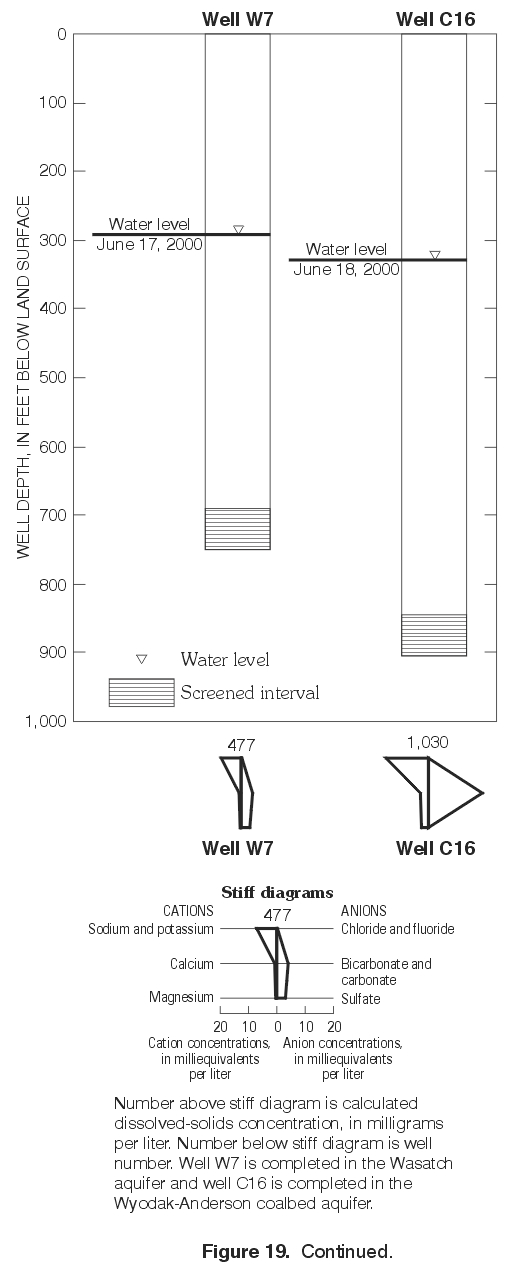
Figure 19. Continued. (Click on image for a larger version, 120 kb) |
Qualitative and statistical examination of the major-ion chemistry of ground-water samples collected during this study suggests there is a relationship between depth and ionic composition in the Wasatch aquifer overlying the coalbed aquifers. A relationship between well depth and major-ion composition of ground water from lower Tertiary aquifers in the Powder River Basin has been noted by many other investigators. The relationship is reflected by the presence of two major-ion compositional systems—generally, a shallow system with mixed cation composition (calcium, magnesium, and lesser amounts of sodium) and either bicarbonate or sulfate as the dominant anion, and a deeper, underlying system where sodium and bicarbonate are the dominant ions. This apparent "zonation" in major-ion composition was first noted by Whitcomb and others (1966) for ground-water samples collected from the Fort Union Formation in Johnson County, Wyoming; the investigators reported that "the quality of water in the Fort Union Formation seems to be related to the depth of the water-bearing zone" (Whitcomb and others, 1966, p. 69). They noted that water from wells less than 500 feet deep in Johnson County, Wyoming, is typically of a sodium-sulfate, calcium-sodium sulfate, or calcium-sulfate type with high dissolved-solids concentrations, and water from deeper wells is sodium-bicarbonate type with lower dissolved solids.
In a study of the geochemistry of waters in the Wasatch and Fort Union Formations in the Powder River Basin within Wyoming, Hagmaier (1971) noted changes in major-ion chemistry with depth in lower Tertiary aquifers and suggested three "divisions" or "zones" with different water types-"shallow ground water, deep ground water from the Wasatch Formation, and deep ground water from the Fort Union Formation" (Hagmaier, 1971, p. 6). In Hagmaier's classification, shallow ground water is typically represented by calcium-sodium-sulfate-bicarbonate-type water in recharge areas and sodium-calcium-bicarbonate-sulfate-type water in local discharge areas; deep ground water from the Wasatch Formation is typically represented by sodium-calcium-sulfate-bicarbonate-type water; and deep ground water from the Fort Union Formation is typically represented by a sodium-bicarbonate-type water. Similarly, a study conducted by the Ground-Water Subgroup (1974) suggested that water from shallow aquifers in the Wasatch and Fort Union Formations in the Gillette area is generally a "calcium and/or magnesium-sulfate water" and "as depth to the aquifer increases, calcium and magnesium ions give way to sodium ions, and sulfate is at least partially replaced by bicarbonate ions," resulting in a sodium-bicarbonate-type water (Ground-Water Subgroup, 1974, p. 35). Lee (1981), in a comprehensive evaluation of the geochemistry of waters in the Fort Union Formation of the Powder River Basin in southern Montana, noted the presence of two distinct geochemical systems—a shallow, localized system generally less than about 200 ft deep that is chemically dynamic with highly variable water chemistry, and a deeper, regional system greater than about 200 ft deep that is chemically stagnant with water chemistry characterized by sodium and bicarbonate. The shallow geochemical system probably coincides with local ground-water flow, while the deeper geochemical system probably is dominated by regional ground-water flow (Lee, 1981; Slagle and others, 1985). Feathers and others (1981) also noted that ionic composition in waters from the Wasatch and Fort Union Formations in the Powder River Basin in Wyoming have a relationship to well depth. Lowry, Wilson, and others (1986, p. 99, fig. 8.4.3-2) noted the same compositional change for waters from lower Tertiary and Upper Cretaceous aquifers in the Powder River drainage basin in Wyoming and Montana occurs and at a depth similar to that noted by Lee (about 200 feet). Rankl and Lowry (1990) examined major-ion composition with depth in lower Tertiary and Upper Cretaceous aquifers in the Powder River Basin in Wyoming and Montana and noted the same changes in ionic composition, although at a depth up to 500 feet (same as Whitcomb and others (1966)). Conceptually, and using nomenclature similar to that presented by Lee (1981), the relation of water type to aquifers in the study area is shown in figure 20.
| Although some of these investigators were often discussing water-quality characteristics and geochemical processes in the Fort Union Formation, or combining the Wasatch and Fort Union Formations into a single hydrogeologic unit in the Powder River Basin, the Wasatch Formation, as discussed earlier, is very similar to and essentially indistinguishable from the Fort Union Formation in depositional and lithological composition (Lowry and Cummings, 1966; Davis, 1976; Flores and others, 1999), although it has been suggested that coal beds and sandstone deposits may be less common in the Wasatch Formation (Lowry and Cummings, 1966, p. 37). Consequently, many of these investigators have noted that the same geochemical processes probably control water chemistry in both lower Tertiary formations (Lowry and Cummings, 1966; Ground-Water Subgroup, 1974; Feathers and others, 1981; Lowry, Wilson, and others, 1986; Rankl and Lowry, 1990). It also should be noted that many of these discussions are referring to the entire Fort Union Formation or combined Fort Union and Wasatch Formations and that processes may differ or be somewhat altered within a single coal bed or zone. | 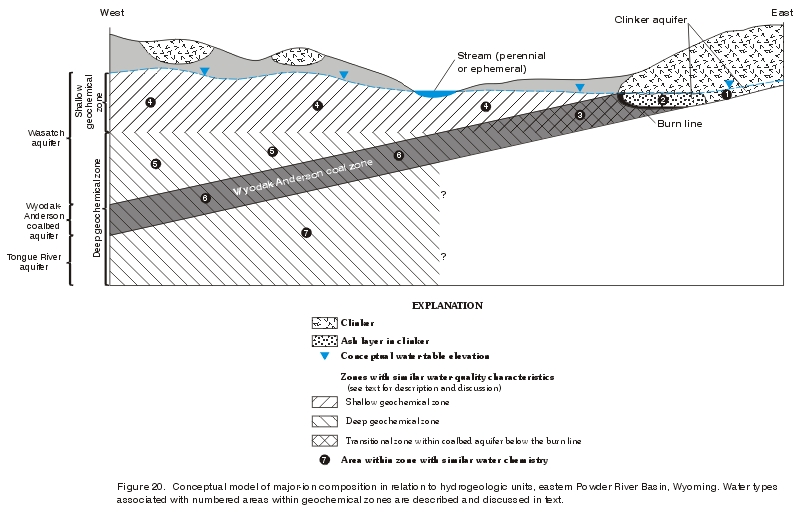
Figure 20. Conceptual model of major-ion composition in relation to hydrogeologic units, eastern Powder River Basin, Wyoming. Water types associated with numbered areas within geochemical zones are described and discussed in text. (Click on image for a larger version, 163 kb) |
Cation exchange is the chemical reaction frequently cited to explain the high percentage of sodium compared to calcium and magnesium in water from the Fort Union Formation in the Powder River Basin (Renick, 1924; Riffenburg, 1925; Lowry and Cummings, 1966; Whitcomb and others, 1966; Hagmaier, 1971; Lee, 1981; Woessner and others, 1981; Lowry, Wilson, and others, 1986; VanVoast and Reiten, 1988; VanVoast, 1991) in Wyoming and Montana, and correspondingly in the Wasatch Formation in Wyoming (Lowry and Cummings, 1966; Whitcomb and others, 1966; Hagmaier, 1971). Cation exchange is a reaction in which the calcium and magnesium in the water are exchanged for sodium that was adsorbed to aquifer solids such as clay minerals, resulting in higher sodium concentrations and softer water (decreased calcium and magnesium concentrations). The generalized reactions (Hem, 1985, p. 13) are as follows:
Na2X + Ca2+ ![]() CaX + 2Na+
(2)
CaX + 2Na+
(2)
Na2X + Mg2+ ![]() MgX + 2Na+ (3)
MgX + 2Na+ (3)
Where Na+ = sodium ion
Ca2+ = calcium ion
Mg2+ = magnesium ion
X = aquifer solid
Hagmaier (1971) and Lee (1981) both noted that calcium and magnesium cations change from dominant to subordinate cations in relation to total cations with increasing depth and as ground water flows away from sources of recharge. Similar conclusions were reached earlier by Hamilton (1970) in western North Dakota and Riffenburg (1925) in the Northern Great Plains of the United States.
Sulfate reduction in water in the Fort Union Formation in Wyoming and Montana was suggested by investigators based on the relatively high percentage of bicarbonate in comparison to sulfate and by the detection of hydrogen sulfide and hydrocarbon gases such as methane in the water at many locations (Riffenburg, 1925; Lowry and Cummings, 1966; Whitcomb and others, 1966; Hagmaier, 1971; Dockins and others, 1980; Lee, 1981; Woessner and others, 1981; Lowry, Wilson, and others, 1986; VanVoast and Reiten, 1988; VanVoast, 1991). These observations, in combination with geochemical modeling at two coal mines, led Martin and others (1988) to conclude that sulfate reduction is likely an ongoing process at some locations in the Powder River Basin. Sulfate reduction is a process where bacteria obtain energy for metabolism from oxidation of relatively simple organic compounds. That oxidation is coupled to sulfate reduction. Byproducts of this activity are bicarbonate and hydrogen sulfide. This process generally occurs when no free oxygen is available (anaerobic or reducing conditions). The generalized reaction (Drever, 1997, p. 161) for sulfate reduction is as follows:
SO2-4 + 2Corganic + 2H2O = H2S + 2HCO3- (4)
The presence of hydrogen sulfide (H2S) is often indicated by the presence of a "rotten-egg" odor; this odor was frequently detected in ground-water samples collected from the Wasatch aquifer and coalbed aquifers during this study. Dockins and others (1980), using stable isotopes of sulfur, showed that bacterially-mediated sulfate reduction is probably responsible for low sulfate concentrations and most of the sulfide production in the Fort Union Formation in the Powder River Basin in southeastern Montana. The investigators also noted that sulfate-reducing bacteria were detected in 25 of 26 of their ground-water samples.
Most of these investigators suggest both processes in combination may explain the observed ionic composition and zonation of water from the Wasatch and Fort Union aquifers in the Powder River Basin of both Wyoming and Montana. The general underlying assumption is that ground water associated with recharge (probably areas 1, 2, and 3 in the Wyodak-Anderson coal zone and area 4 in the shallow geochemical zone, fig. 20) is represented by water dominant in calcium, magnesium, and sulfate, with lesser amounts of sodium and bicarbonate (Hagmaier, 1971; Lee, 1981; Woessner and others, 1981). As ground water flows away from the source of recharge, the interaction between water and rock increases. Sodic lithologic units are encountered as the ground water moves along a flowpath and calcium and magnesium ions are exchanged for sodium ions attached to aquifer solids. Anaerobic sulfate-reducing bacteria, in the presence of organic material in coals and carbonaceous shales, also act on the ground water as it moves along a flowpath. Therefore, both reactions result in a decrease in calcium, magnesium, and sulfate, and a corresponding increase in sodium and bicarbonate as ground water flows away from the source of recharge and results in a water that evolves to a sodium-bicarbonate type (areas 5 and 7 in the deep geochemical zone and area 6 in the Wyodak-Anderson coal zone, fig. 20).
With the possible exception of Hagmaier (1971), Lee's evaluation of the water quality in the Fort Union Formation in the Powder River Basin in southern Montana is still the most comprehensive examination of the water chemistry in relation to hydrogeologic and geochemical conditions. However, it should be noted that many of the conclusions reached by Lee (1981) are very similar to conclusions reached by Hagmaier (1971) for the geochemistry of waters in the same aquifers in the Powder River Basin in Wyoming, even though both investigators appear to have reached their conclusions independently. As discussed previously, Lee suggests the presence of two distinct geochemical systems: a shallow, localized, chemically dynamic system generally less than 200 feet (area 4 in the shallow geochemical zone, fig. 20) and an underlying deeper, regional, chemically static system (areas 5 and 7 in the deep geochemical zone and area 6 in the Wyodak-Anderson coal zone, fig. 20). At topographically high locations and from springs, water from the shallow system probably represents recharge waters and the dominant ions are magnesium, calcium, sodium, and bicarbonate, with moderate amounts of sulfate and low concentrations of chloride. At topographically low locations, water in the shallow system is dominant either in sodium and sulfate with lower concentrations of magnesium, calcium, and bicarbonate, or is dominant in sodium and bicarbonate with lower concentrations of magnesium, calcium, and sulfate.
Lee proposed three general trends in the major-ion composition of ground water as it flows away from recharge areas as well as the geochemical processes that might produce the trends. The three trends are: (1) enrichment in sodium and sulfate, (2) enrichment in sodium and depletion of sulfate, and (3) depletion of sodium and sulfate. Lee found the first trend was the most common trend in the shallow system, although all three trends were observed. Lee concluded that the primary chemical reactions that contribute to sodium enrichment are dissolution of minerals such as sodium feldspars by infiltrating recharge waters in combination with cation-exchange on clay minerals, although other reactions of less significance also may occur. Direct sulfate enrichment may be attributed to the weathering of pyrite and dissolution of gypsum (also noted in Dockins and others (1980)), or indirectly by precipitation of calcium and magnesium carbonates that remove bicarbonate from solution (also suggested by Hagmaier (1971)). As ground water continues to flow away from the source of recharge, the trend towards sulfate enrichment is reversed and relative depletion of sulfate is commonly observed (second trend).
Bacterially mediated sulfate reduction is the chemical reaction suspected to explain the decreases in sulfate in both the shallow and deeper systems. Lee proposed that the observed trend of sodium and sulfate depletion (third trend) may be explained by mixing of infiltrating ground water with older ground water that results in dilution. In addition, water in the deeper system, typically with sodium-bicarbonate-type waters, may locally leak upward into the shallow system and decrease the relative percentage of sulfate. In the chemically stagnant deeper system, Lee suggests bacterially mediated sulfate reduction was the dominant geochemical process and results in water that is primarily sodium-bicarbonate type.
Carbonate equilibria also may be controlling the amount of dissolved calcium and magnesium in solution. The presence of dissolved constituents at relatively high concentrations (i.e., sodium and bicarbonate concentrations in most samples collected from all aquifers in the study area) can control the amount of calcium and magnesium in solution through the precipitation of carbonate minerals. The reader is referred to Hem (1985), Stumm and Morgan (1996), and Drever (1997) for more detailed discussions of carbonate equilibria and equilibrium water chemistry.
Waters in the coalbed aquifers in some areas may evolve to a sodium-bicarbonate-type water through a different combination of hydrological and geochemical processes. Heffern and Coates (1999) examined major-ion composition in clinker associated with the Wyodak-Anderson coalbed aquifer along the eastern margin of the basin and study area (Rochelle Hills area) and speculated on the processes responsible for the composition of the waters in the deposits. As described earlier, the clinker in this area is often directly updip and in hydraulic connection with the Wyodak-Anderson coalbed aquifer (fig. 20) and the deposits in this area are suspected to be the source of much of the recharge to the aquifer in the study area. The investigators noted several trends in major-ion chemistry in the clinker: (1) Waters upgradient of the burn line (the contact between the clinker and unburned coal or overburden) are highly oxygenated with low dissolved-solids concentrations and are primarily calcium-bicarbonate-type waters (area 1 in the Wyodak-Anderson coal zone, fig. 20), and (2) waters "ponded" adjacent to or near the burn line are frequently characterized by high sulfate concentrations and high dissolved-solids concentrations (area 2 in the Wyodak-Anderson coal zone, fig. 20). Waters in the Wyodak-Anderson coalbed aquifer immediately downgradient of the burn line (and in hydraulic connection with the clinker) are frequently characterized by moderate dissolved-solids concentrations and sodium-bicarbonate-type waters typically seen in the coalbed aquifers in the study area (areas 3 and 6 in the Wyodak-Anderson coal zone, fig. 20). The investigators speculate on the processes that result in the observed chemistry. The following quotation is from Heffern and Coates (1999, p. 249) and explains their hypothesis:
Mine permits show that wells drilled into ponded clinker water near this contact commonly have high levels of sulfate and TDS. We postulate that these high concentrations are due to dissolution of: 1) sulfates and pyrite in the coal near the burn line, and 2) minerals in the ash layer at the base of the clinker, as highly oxygenated ground water flows downdip through the clinker towards the coal. As this water moves downdip into and through the coal, calcium and sulfate precipitate out in fractures as gypsum, and magnesium and sulfate as epsomite (Davis, 1976). Water in the coal, and in clinker downdip from the coal, is typically of the sodium-bicarbonate type, with moderate TDS levels.
Coalbed methane in the Powder River Basin is believed to be biological in
origin (biogenic) (Law and others, 1991; Rice, 1993; Gorody, 1999), a product of
anaerobic bacterial decomposition of organic matter through a series of redox
reactions. In samples collected from the coalbed aquifers during this study, the
high percentage of sulfate nondetections (greater than 50 percent) and low
sulfate concentrations when detected is suggestive of waters that have undergone
or are undergoing sulfate reduction. VanVoast (1991, p. 1,143) noted that
methane encountered in the Powder River Basin "invariably coincides with
conditions of chemically reduced groundwater containing Na and HCO3
as the principal ions." In addition, the author noted that "a chemical
model accounting for methane and the associated water-quality conditions
requires the exchange of cations Ca and Mg (to clays) for Na (to water) followed
by: 4CH2O (coal) + SO4 + H2O ![]() H2S +3HCO3 + CH4" (VanVoast, 1991, p.
1,143). Rice and others (2000) also observed similar low concentrations and
nondetections of sulfate in ground-water samples collected from 47 wells
completed in the coalbed aquifers; similarly, the investigators noted that the
low sulfate concentrations "are consistent with water in contact with a
coal reservoir that has undergone or is undergoing methanogenesis" (Rice
and others, 2000, p. 5).
H2S +3HCO3 + CH4" (VanVoast, 1991, p.
1,143). Rice and others (2000) also observed similar low concentrations and
nondetections of sulfate in ground-water samples collected from 47 wells
completed in the coalbed aquifers; similarly, the investigators noted that the
low sulfate concentrations "are consistent with water in contact with a
coal reservoir that has undergone or is undergoing methanogenesis" (Rice
and others, 2000, p. 5).
Methanogenesis is another type of redox chemical reaction. The anaerobic bacterial decomposition of organic matter occurs as a series of redox chemical reactions—each series or stage of redox reactions occur in a specific order (succession) and are mediated by different types of bacteria (Chapelle and others, 1993; Chapelle, 2001); this series of reactions essentially represents a microbial "food chain" where different types of bacteria utilize relatively simple organic matter or compounds as a source of energy (Chapelle and others, 1993; Chapelle, 2001). Sulfate reduction (described earlier) is one of these reactions. After all or most sulfate has been consumed through the process of sulfate reduction, the anaerobic bacterial decomposition of organic matter occurs in three stages and each stage or reaction is mediated by a different type of bacteria—the final stage or process is known as methanogenesis (Rice, 1993) and the bacteria that mediate the process are known as "methanogens." Although these redox chemical reactions occur in successive stages, both sulfate reduction and methanogenesis can exist within separate zones of the same aquifer (Chapelle and others, 1993; Chapelle, 2001). The methanogens utilize relatively simple organic compounds (produced in two prior stages of anaerobic decomposition by the breakdown of complex organic matter by acetogenic and fermentative bacteria such as hydrogen (H2), carbon dioxide (CO2), acetate, formate, and simple alcohols, to produce methane). There are two metabolic pathways used by bacteria to produce methane after sulfate reduction has ended—methyl-group fermentation and reduction of carbon dioxide (Schoell, 1980; Wolternate and others, 1984; Jenden and Kaplan, 1986; Whiticar and others, 1986). The two generalized reactions (Chapelle, 2001) showing the metabolic pathways for bacterial conversion of simple organic matter into methane are as follows:
Methyl-group fermentation CH3COOH
![]() CH4 + CO2
(5)
CH4 + CO2
(5)
acetic acid
Carbon dioxide reduction CO2 + 4H2
![]() CH4 + 2H2O
(6)
CH4 + 2H2O
(6)
Rice (1993) examined stable isotopes of gas and water and suggested that the second reaction, carbon dioxide reduction, was the metabolic pathway responsible for most (about 80 percent) of the biogenic generation of methane in coal beds of the Powder River Basin and methyl-group fermentation was responsible for the rest. Similarly, Gorody (1999) also examined the isotopic composition of gas and water from the coal beds and also concluded that reduction of carbon dioxide by methanogenic bacteria rather than methyl-group fermentation is probably responsible for most of the large quantities of methane present in the Wyodak-Anderson coal zone in the Powder River Basin. Rice (1993) suggests that biogenic methane generation in the Powder River Basin occurred between 10 and 35 million years ago, the time of estimated maximum burial of the coal beds. In contrast, Gorody (1999) suggests that methane generation may have continued to at least the Pleistocene age.
In the past, both sulfate reduction and methanogenesis have affected the chemistry of waters in the Wasatch aquifer and coalbed aquifers. On the basis of work in this and other studies described herein, sulfate reduction was likely the dominant geochemical process responsible for the sodium-bicarbonate-type waters in the deep aquifers in the study area. The low to nondetectable concentrations of sulfate in coalbed aquifers in the study area documented in this study and in Rice and others (2000), in combination with earlier work by Dockins and others (1980) and other studies described earlier, suggests that sulfate reduction may be an ongoing process in some locations in aquifers in the study area. Presumably, the sulfate reduction is occurring primarily in the Wasatch aquifer overlying the coalbed aquifers (except possibly along eastern basin margin—see discussion in Heffern and Coates (1999) and discussion herein) because the near absence of sulfate in the coalbed aquifers indicates the waters in the coalbed aquifers are past sulfate reduction in terms of terminal electron-accepting processes (see discussion in Chapelle (2001)). Because of the absence of sulfate, waters in the coalbed aquifers can undergo methanogenesis if the organic precursors necessary for activity by methanogenic bacteria are present in the aquifers. Work to answer these questions is well outside the scope of this study. Future work will hopefully determine the location and extent of active sulfate reduction and determine if methanogenesis in the aquifers is occuring in any of the study area and throughout the Powder River Basin.
Results from this and previous studies suggest that reducing conditions exist in significant portions of the Wasatch and coalbed aquifers. Very reducing conditions can affect the chemistry (i.e., mobility) of many other constituents, particularly redox-sensitive species (Hem, 1985; Stumm and Morgan, 1996; Drever, 1997). Iron and manganese are detected at relatively high concentrations in the Wasatch aquifer and coalbed aquifers, often at concentrations greater than regulatory standards (Martin and others, 1988; Rice and others, 2000). Both iron and manganese are highly sensitive to redox conditions and both are more soluble in reducing conditions (Hem, 1985).
Low to nondetectable concentrations of many trace elements in waters from the coalbed aquifers in the Powder River Basin in Wyoming have been reported by other investigators (Drever and others, 1977; Martin and others, 1988; Rice and others, 2000); when detected, concentrations of many trace elements are well below any applicable USEPA regulatory standards. Hydrogen sulfide (H2S) produced by sulfate reduction can strongly affect (decrease) the solubility of many metals (Drever, 1997). In addition, the low to nondetectable sulfate concentrations in the coalbed aquifers can influence the solubility of trace elements such as barium. Rice and others (2000) noted that barium is frequently detected in the coalbed aquifers at concentrations higher than typically seen for most ground waters. The solubility of barium is primarily controlled by barite (barium sulfate) (Hem, 1985), so the absence of sulfate may be responsible for the relatively high concentrations observed in the waters.
Isotopes are atoms of the same element that have the same numbers of protons and electrons but different numbers of neutrons. The difference in the number of neutrons between different isotopes of the same element means they have the same atomic number but different atomic masses. Environmental isotopes are naturally occurring isotopes of elements commonly found in the natural environment (typically hydrogen, carbon, nitrogen, oxygen, and sulfur). Isotopes are called stable if not involved in any natural radioactive decay or called radioactive if they undergo radioactive decay. Stable isotopes are typically used in hydrogeology as "tracers of water, carbon, nutrient, and solute cycling" (Clark and Fritz, 1997, p. 5) whereas radioactive isotopes are typically used "to estimate the age or circulation of ground-water" (Clark and Fritz, 1997, p. 5). Both radioactive and stable isotopes were examined during this study.
Tritium (3H) is a radioactive isotope of hydrogen (H) commonly used as a tool to provide an approximate age for ground water. Tritium atoms are unstable and undergo radioactive decay with a half-life of 12.43 years (Plummer and others, 1993). Unlike other radioisotopes, tritium atoms can substitute for other hydrogen atoms in the water molecule. The incorporation of tritium directly into the water molecule makes tritium an excellent tool for qualitatively age-dating ground water. Tritium is produced by cosmic radiation naturally in the upper atmosphere at very low concentrations and is incorporated into precipitation. However, atmospheric testing of thermonuclear devices beginning in the early 1950's and continuing through the early 1960's increased tritium concentrations by several orders of magnitude above natural levels, resulting in a global atmospheric spike in tritium concentrations measured in precipitation. The atmospheric spike occurred during and after thermonuclear device testing and "created a tritium reservoir in the stratosphere which contaminated global precipitation systems for over four decades" (Clark and Fritz, 1997, p. 177). A test-ban treaty was signed in 1963, effectively ending large-scale atmospheric testing of thermonuclear devices and the resulting large increases in atmospheric tritium. Non-signatory nations (France and China) conducted small atmospheric tests of thermonuclear devices until 1980 (Clark and Fritz, 1997) but these tests probably contributed little to tritium concentrations in precipitation (Michel, 1989). Atmospheric concentrations decreased after the test-ban treaty by decay in the atmosphere and loss to the hydrologic cycle (primarily the oceans). Tritium concentrations in precipitation have only recently (about 1990) approached natural levels. Concentrations probably will never completely return to natural levels because low concentrations of tritium continue to be released by other anthropogenic sources such as nuclear power plants and weapons plants (Clark and Fritz, 1997).
Tritium in ground water is primarily from precipitation that infiltrated downward to the aquifer (ground-water recharge). Recharge to ground water originates as precipitation, therefore, increased levels of tritium present in precipitation during and after nuclear device testing can be measured in ground-water samples to qualitatively estimate the time that the water entered the aquifer and became isolated from the atmospheric source of tritium. Tritium is generally used as a qualitative dating tool because tritium concentrations have varied considerably, both spatially and temporally (Michel, 1989). The presence of elevated levels of tritium in ground water is considered an indicator of recent or active ground-water recharge that has occurred since the early 1950's and has become the standard for defining "modern" ground water (Plummer and others, 1993; Clark and Fritz, 1997). Because modern ground water is defined by the very presence of tritium, water with detectable concentrations also is frequently referred to as post-bomb water or ground water with some component of recharge after 1952. Ground water with little or no detectable tritium is defined as "submodern" or older and often is referred to as pre-bomb water or ground water recharged prior to 1952.
Tritium concentrations are expressed in picocuries per liter (pCi/L) or tritium units (TU) where 1 TU is equal to 1 3H atom in 1018 atoms of H, or 1 TU is equal to about 3.24 pCi/L (Plummer and others, 1993). From a practical standpoint, tritium concentrations in ground water recharged prior to nuclear device testing (submodern or pre-bomb water) would be less than about 0.8 TU or 2.6 pCi/L; greater concentrations would indicate modern water that has been recharged after atmospheric thermonuclear device testing began (modern or post-bomb water) or ground water that is a mixture of submodern and modern water (Plummer and others, 1993; Clark and Fritz, 1997).
Concentrations detected in the water samples discussed in this report are compared to qualitative interpretations of tritium concentrations in ground water for continental regions as presented in Clark and Fritz (1997, p. 185), and reproduced here in its entirety for convenience:
< 0.8 TU Submodern—recharged prior to 1952
0.8 to ~ 4 TU Mixture between submodern and recent recharge
5 to 15 TU Modern (< 5 to 10 years)
15 to 30 TU Some "bomb" tritium present
> 30 TU Considerable component of recharge from the 1960's or 1970's
> 50 TU Dominantly the 1960's recharge
Tritium concentrations in ground-water samples collected from springs, monitoring wells, and coalbed methane production wells as part of this study are presented in table 10; for convenience, concentrations are presented in both pCi/L and TU.
Tritium concentrations in water samples collected from spring S1 (about 24 TU) and spring S2 (about 16 TU) suggest that both were recharged after 1952 and contain modern or post-bomb water (table 10). The tritium concentrations are both within the "some bomb tritium present" category presented by Clark and Fritz (1997), indicating that both springs were recharged after 1952 and are likely discharging modern or post-bomb water. The presence of modern water in both springs is indicative of recent recharge; both springs had calcium and magnesium as dominant cations and the presence of modern water confirmed suggestions by other investigators (discussed earlier) that these two cations would be dominant in recharge waters in the study area.
Ground-water samples were collected from eight monitoring wells completed in the Wasatch aquifer to examine tritium concentrations in aquifers overlying the coalbed aquifers (table 10). Tritium was not detected in two of eight monitoring wells completed in the Wasatch aquifer. Detectable tritium concentrations in the remaining six wells ranged from the MRL (about 0.3 TU or 1 pCi/L) to about 2.8 TU. Therefore, tritium concentrations in six of eight water samples collected from wells completed in the Wasatch aquifer overlying the coalbed aquifers suggest the water is submodern or pre-bomb (tritium concentration less than 0.8 TU). Tritium concentrations in the remaining two samples are in the category (0.8 to 4 TU) that suggests a mixture between submodern (pre-bomb) and modern (post-bomb) water, although the low concentrations suggest that ground water in these wells have very little modern water.
Tritium was not detected in eight of thirteen ground-water samples collected from monitoring wells and coalbed methane production wells completed in coalbed aquifers (table 10). Of the five wells with detectable concentrations, tritium was detected at the MRL (about 0.3 TU) in four wells and at a concentration of 0.6 TU in one well. The absence of any tritium at concentrations above 0.8 TU suggests that ground water in the coalbed aquifers probably is submodern or pre-bomb, indicating that no recharge water is likely to have entered the aquifers and traveled to the vicinity of the sampled wells since at least the early 1950's.
Ratios of the stable isotopes of oxygen (oxygen-18/ oxygen-16, or 18O /16O) and hydrogen (deuterium/ hydrogen, or 2H/H) in the water samples were examined in relation to meteoric water lines, to vertical distribution in monitoring-well clusters, and to spatial distribution. Hydrogen-2 (deuterium or 2H) is a stable heavy isotope of hydrogen containing an extra neutron, and oxygen-18 (18O) is a stable heavy isotope of oxygen containing two extra neutrons. The variation in the number of neutrons in an element results in a different mass. This difference in mass causes fractionation to occur by a process known as Rayleigh fractionation (Clark and Fritz, 1997). Isotopic data are reported as ratios because isotopic ratios are determined more precisely than actual abundances. The isotopic concentration or ratio is expressed as the difference between the measured ratios of the sample and the reference water divided by the ratio of the measured reference water. Vienna Standard Mean Ocean Water (VSMOW) is generally used as the standard reference water for d18O and d2H. The ratio, commonly known as delta (d), is expressed in units of parts per thousand or per mil (‰) because the differences are very small. For example, a sample with a d18O of -20 ‰ is depleted in d18O by 2 percent, or 20 ‰ relative to the standard. This ratio is calculated as follows (example for d18O):
d18O sample (in ‰) = (7)
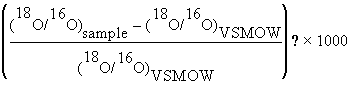
An equation of the same form is used to calculate δ2H. The analytical precision for d18O values in a given laboratory is usually about +/- 0.2 ‰ and for d2H, the analytical precision is usually +/- 1.0 ‰ (Clark and Fritz, 1997). In this study, a field replicate collected at well C15 had a difference of 0.7 ‰ for d2H and 0.05 ‰ for d18O between the normal environmental sample and the field replicate (table 11).
The values of d18O and d2H in precipitation become progressively lighter from the equator towards the poles, from lower to higher elevations, and with the distance inland from the ocean. When paired d18O and d2H values of precipitation throughout the world are plotted, they fall closely to a straight line known as the global meteoric water line (GMWL) (Craig, 1961) which is defined as follows:
d2H = 8 d18O + 10 (8)
Similarly, paired d18O and d2H values for North American continental precipitation have been plotted and they also fall closely to a straight line that has the same slope but is shifted downward from GMWL (Gat, 1980).
d2H and δ18O values in ground water are representative of values in precipitation that recharge the ground water, unless some process after the water reaches the earth's surface as precipitation causes isotopic fractionation, and consequently, deviation from meteoric water lines. Some processes that cause fractionation are evaporation, exchange with the aquifer matrix, or recharge that occurred at a different temperature or under a different climate. Local meteoric water lines also may deviate from the GMWL. Shifts in d18O values may be affected by interaction with the aquifer matrix. Calcite interactions in carbonate aquifers or more complex exchanges with the rock matrix in geothermal systems are commonly observed causes of shifts in the d18O values. In general, there are fewer shifts in the d2H in aquifer systems because fifty percent or more of the total oxygen in the system is usually resident in the rocks, but almost all of the hydrogen in the system is in the water (Drever, 1997). Coalbed aquifers are exceptions to that general rule, because significant amounts of hydrogen are tied up in the coal. In addition, interaction between gases and water may introduce a more complex model, particularly when methanogenesis is present. As described earlier, the biogenic production of methane is a complex process of a mixed bacterial population and results in the production of carbon dioxide and hydrogen gases as well as methane. d2H values have been observed to shift in water samples collected from landfills because of active methane production (Siegel and others, 1990; Hackley and others, 1996; Clark and Fritz, 1997).
| Collection of d2H and δ18O in precipitation was not possible as part of this study and no local values were available; therefore, the d2H and δ18O values in ground-water samples (table 11) were plotted and compared to published values for precipitation (fig. 21). The values plotted close to the GMWL, the meteoric water line for North American continental precipitation, and a local meteoric water line estimated by Gorody (1999) for the Powder River Basin. The close proximity to the meteoric water lines, including samples from two springs with modern water, suggests that the water was of meteoric origin, in contrast to water present in the aquifer materials during deposition (connate). The samples also plotted in the more negative area (i.e., left side) of the graph, which is appropriate for waters recharged at colder temperatures or in a colder climate, mid-latitudes, and mid-continent. Many of the values plotted slightly below all three meteoric water lines, despite different aquifers of origin, suggesting some isotopic fractionation, possibly by evaporation. The water samples were intermixed on the plot and did not group into separate areas of the graph based on the aquifer of origin. This could indicate that there was intermixing of the waters in the aquifers or it could indicate that the different aquifers were subject to similar recharge and or evolutional paths for the water, so that the net difference in the d2 and d18O values was minimal. Similar d2H and d18O values were observed in four ground-water samples collected from the Wyodak-Anderson coalbed aquifer at two coal mines in the southern part of the Powder River Basin (Martin and others, 1988). | 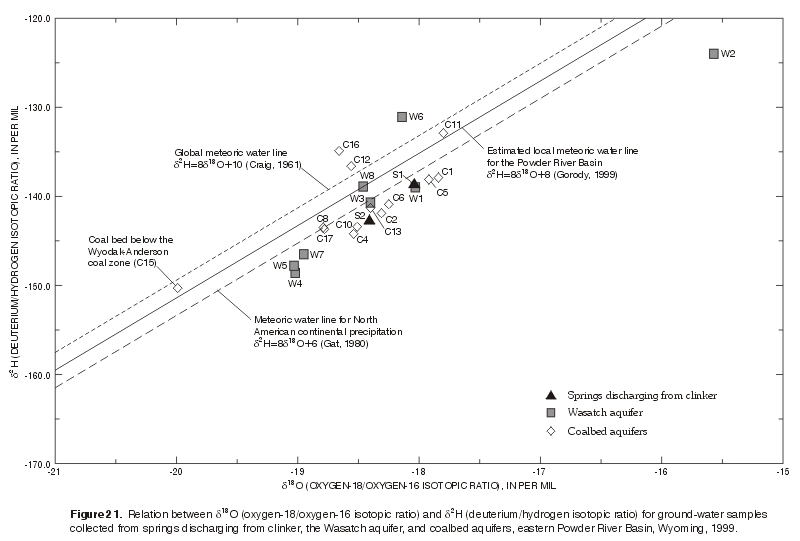
Figure 21. Relation between δ18O (oxygen-18/oxygen-16 isotopic ratio) and d2H (deuterium/hydrogen isotopic ratio) for ground-water samples collected from springs discharging from clinker, the Wasatch aquifer, and coalbed aquifers, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 106 kb) |
d2H and d18O values were examined in the vertical dimension at the three monitoring-well clusters where major-ion chemistry was previously discussed. In the monitoring-well cluster composed of wells W5, W4, and W3, and nearby coalbed methane production well C12, depth to water increased with the depth of the well (fig. 22). The water level in the coalbed well was measured before the well was pumped to remove water for gas production, while the water levels in the wells completed in the Wasatch aquifer were collected after pumping of the coalbed methane production field began. The water level in the coalbed well (C12) probably had drawn down farther by 1999 than is presented in figure 22 because this well was a production well that produced water and gas in 1999. Hydraulic head (fig. 22) decreased with depth and indicated a hydraulic potential for downward ground-water flow if geologic conditions allow.

Figure 22. d2H (deuterium/hydrogen isotopic ratio), d18O (oxygen-18/oxygen-16 isotopic ratio), and major-ion chemistry in relation to the clustered wells W5, W4, W3, and C12 in the eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 161 kb) |
Figure 22. Continued. (Click on image for a larger version, 157 kb) |
d2H values generally increased (became less negative) with depth (fig. 22). d2H values in the two shallow Wasatch wells (W5and W4) are about the same, although the middle Wasatch well (W4) was slightly more negative than the uppermost well (W5); the difference between the two d2H values is probably within the precision of the d2H analysis. d2H increased dramatically between the two shallow Wasatch wells (W5 and W4) and the deepest Wasatch well (W3). Similarly, d18O values were about the same between the two shallowest Wasatch wells but increased with depth between the middle Wasatch well (W4) and the deepest Wasatch well (W3). Physical and chemical processes such as methanogenesis and hydrogen-sulfide exchange reactions can shift d2H values towards less negative values (deuterium enrichment) without corresponding shifts in d18O values (Hackley and others, 1996, and references therein). If the sandstone lenses screened by the wells were assumed to be hydraulically connected, one of these processes could increase d2H values as ground water moves downward. However, a shift in δ18O is observed between the two shallow Wasatch wells (W5 and W4) and the deep Wasatch well (W3) and suggests that these processes have probably not affected the d2H values between these wells. The shifts observed in δ2H and d18O between the two shallow Wasatch wells and the deepest Wasatch well almost parallels the meteoric water lines (fig. 21) and this type of deviation from the meteoric water lines may be more indicative of differences in recharge temperatures rather than processes such as methanogenesis or hydrogen-sulfide exchange reactions. In addition, it is interesting to note that the shift in d2H between the two shallowest Wasatch wells and the deepest Wasatch well corresponds to a change in water type (fig. 22) and a decrease in sulfate concentrations (figs. 17 and 22).
d2H continued to increase with depth between the deepest Wasatch well (W3) and the underlying Wyodak-Anderson coalbed well (C12) (figs. 17 and 22). Waters in both wells were sodium-bicarbonate type. Sulfate concentrations continued to decrease with depth, with a concentration of about 2,400 mg/L in the deep Wasatch well (W3) and less than the MRL of 0.1 mg/L in the coalbed well (C12). In this case, d2H shifted to a less negative value without a corresponding shift in the d18O values (fig. 22). As mentioned previously, this pattern is sometimes observed where waters have undergone or were undergoing methanogenesis or hydrogen-sulfide exchange reactions and might be observed if the waters in both wells were assumed to be originally isotopically the same, in hydraulic connection, and moving downward. Future work will hopefully determine if either geochemical process affected or currently affects the water chemistry in the deep wells.In the monitoring-well cluster composed of wells W7 (completed in Wasatch aquifer) and C16 (completed in Wyodak-Anderson coalbed aquifer) (fig. 23), d2H increased (became less negative) with depth; d18O also slightly increased (became less negative) with depth, but the difference in the d18O values is probably within the precision of the analysis. Hydraulic head decreased with depth, indicating the potential for downward ground-water flow. Waters in both wells were sodium-bicarbonate type. Sulfate concentrations decreased with depth (fig. 19), with a concentration of about 160 mg/L in the Wasatch well (W7) and about 1 mg/L in the coalbed well (C16). This pattern in stable isotope values, hydraulic gradient, and major-ion chemistry is very similar to the pattern just described for wells W3 and C12 in the monitoring-well cluster.
In contrast, a very different pattern was observed in the monitoring-well cluster composed of wells W8 and C17 (fig. 23). Both d2H and d18O decreased (became more negative) with depth. Unlike the other two monitoring-well clusters, hydraulic head increased with depth, indicating the potential for upward ground-water flow. Major-ion chemistry also was different, as the water from the Wasatch well (W8) immediately overlying the coalbed aquifer was a mixed type, and the water in the coalbed well (C17) was a sodium-bicarbonate type. Sulfate concentrations decreased with depth (fig. 18), with a concentration of about 740 mg/L in the Wasatch well (W8) and less than 1 mg/L in the coalbed well (C17). In the other two monitoring-well clusters, the waters in samples collected from the Wasatch wells immediately overlying the coalbed aquifer were sodium-bicarbonate-type waters, the same as the waters in the underlying coalbed aquifers.
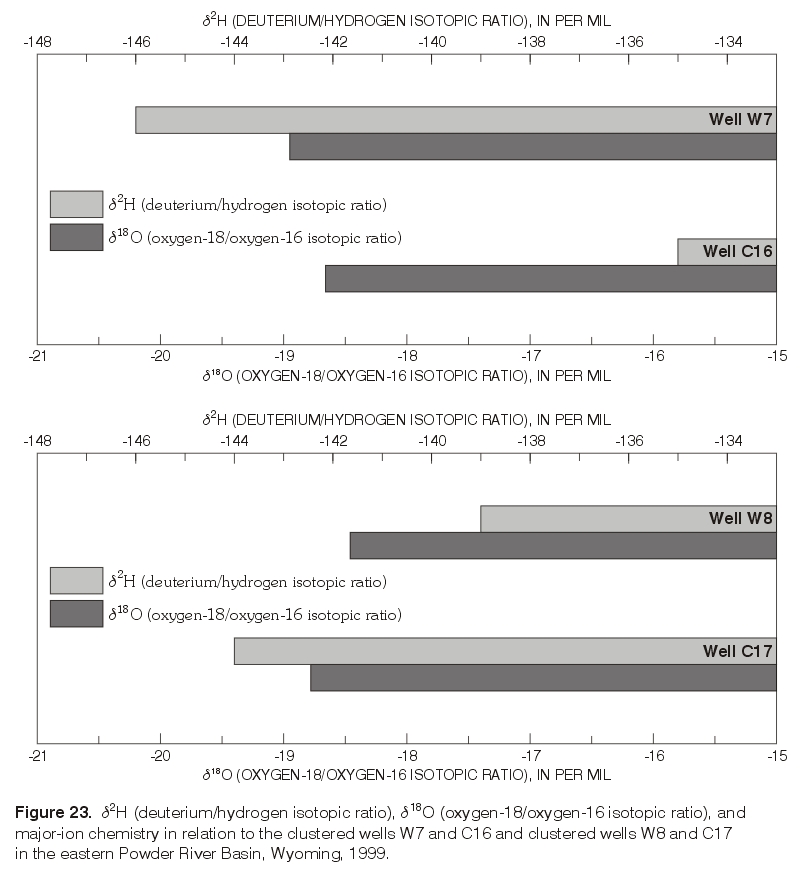
Figure 23. d2H (deuterium/hydrogen isotopic ratio), d18O (oxygen-18/oxygen-16 isotopic ratio), and major-ion chemistry in relation to the clustered wells W7 and C16 and clustered wells W8 and C17 in the eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 157 kb) |
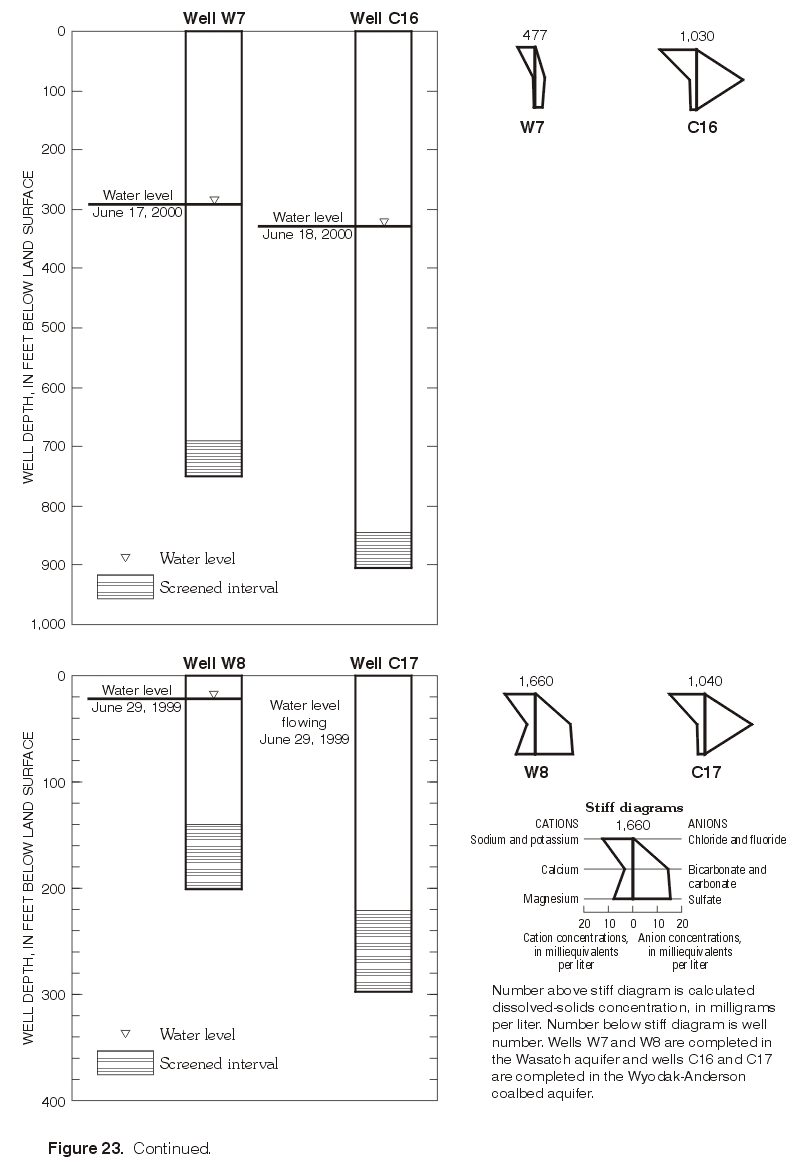
Figure 23. Continued. (Click on image for a larger version, 168 kb) |
Two explanations or hypotheses are proposed to explain the observed changes in stable isotope values and major-ion chemistry with increasing well depth at the three monitoring-well cluster locations. The first hypothesis requires an assumption of hydraulic connection between the intervals screened by successively deeper wells. In this case, the observed changes with depth are the result of geochemical processes (described earlier for the Powder River Basin) as ground water moves downward through the Wasatch Formation into the underlying coalbed aquifer. The second explanation is that there are two different aquifers or aquifer systems with minimal hydraulic connection - (1) the upper Wasatch Formation, represented by the two sandstone lenses screened by the two shallow Wasatch wells (wells W5 and W4) in the first monitoring-well cluster discussed, and (2) the coalbed aquifers and overlying deep Wasatch sandstone lenses, represented by the deep Wasatch well (W3) and coalbed well (C12) in the first monitoring-well cluster discussed, and Wasatch well W7 and coalbed well C16 in the second monitoring-well cluster discussed. At the third monitoring-well cluster examined, the shallow Wasatch well (W8) and underlying coalbed well (C17) would represent the same two different aquifers or aquifer systems. In this second hypothesis, each of the two aquifers or aquifer systems have different major-ion chemistry and stable isotope values because the waters in the different aquifers or aquifer systems are subject to different recharge and/or evolutional paths.
The shallow aquifer or aquifer system would be part of the "shallow geochemical zone" described earlier for the Powder River Basin (and associated geochemical processes, primarily in the context provided by Lee (1981)) while the deeper aquifer or aquifer system would be part of the "deep geochemical zone" described earlier (see fig. 20 and related discussion in earlier section, Major-Ion Chemistry).
d2H values were examined spatially (figs. 24 and 25) for both wells completed in the Wasatch aquifer and the coalbed aquifers. Springs discharging from clinker were included with the wells completed in the Wasatch aquifer. A break in δ2H values can be observed along a northwest to southeast trend with both groups. An inferred -140 ‰ d2H value line is shown in both figures as an arbitrary reference value to examine relative increases or decreases in d2H values. Samples collected from wells completed in the coalbed aquifers, except for the sample from the Big George coal bed, followed a pattern where the more negative d2H values plotted towards the center of the basin, whereas the less negative values plotted near the outcrop area. The pattern for the samples collected from the Wasatch aquifer and the springs was opposite. The values more negative than -140 ‰ plotted near the outcrop area, while the values less negative than -140 ‰ plotted towards the basin center. The d2H -140 ‰ line in the coalbed aquifer and the d2H -140 ‰ line in the overlying Wasatch aquifer spatially were relatively close to each other given the size of the dataset. Further investigation of this possible spatial relation is required. The pattern may be the result of sample size, different recharge mechanisms, or geochemical processes, or the processes producing these differences may be independent.
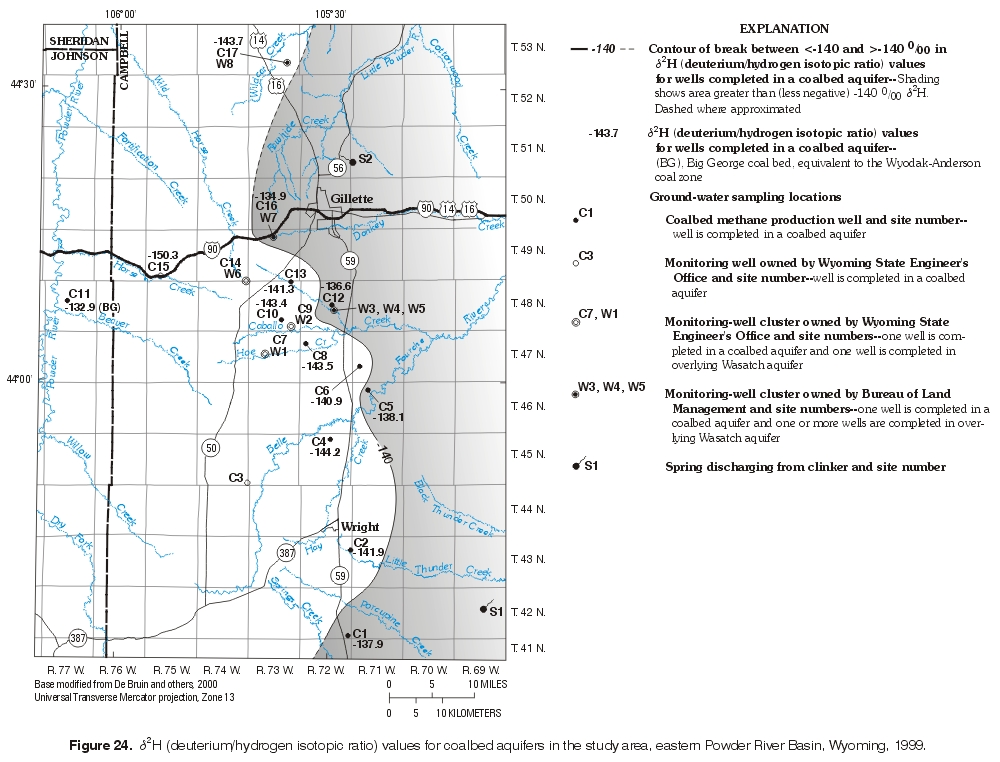
Figure 24. d2H (deuterium/hydrogen isotopic ratio) values for coalbed aquifers in the study area, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 340 kb) |
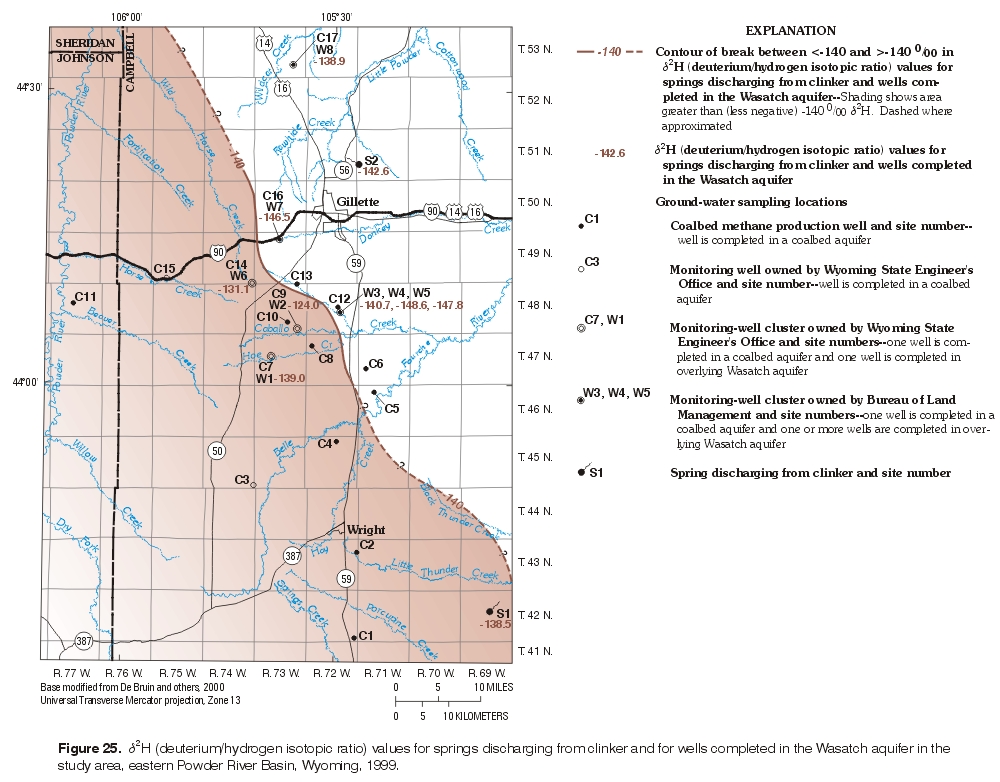
Figure 25. d2H (deuterium/hydrogen isotopic ratio) values for springs discharging from clinker and for wells completed in the Wasatch aquifer in the study area, eastern Powder River Basin, Wyoming, 1999. (Click on image for a larger version, 358 kb) |

{kind=link}
{kind=link}