|
We have been attacking the problem of assessing the fate of heavy metals in coastal sediments over the past 5 years. Our primary goal has been to determine what controls their cycling in the sediment column. With this information, we can then address two important questions that concern environmental policy makers in coastal, urban areas. What is the ultimate fate of toxic metals that were released into the coastal ocean in the past and remain in coastal sediments today? And, what is the fate of metals that are currently being added to the coastal ocean in the treated sewage effluent that is released now? Our study has focused on the reactions of 3 toxic metals (Ag, Cu, and Pb) in sediments and in sedimentary pore waters and on the potential for release of these metals from sediments to overlying water. This work complements the long-term time-series studies of contaminant and sediment transport conducted in coastal Massachusetts by the USGS (Section 7, this report). We are also linked with related studies on oxygen penetration in sediments (Section 8, this report). The results discussed below relate two contrasting sites in the Boston area (Kalnejais, 2005).
Hingham Bay, in Boston Harbor, is a shallow-water site (6 m) where organic matter is oxidized at the very high annual rate of 1200 µmol C/cm2/y. As a consequence of this rapid organic matter oxidation rate, the entire sequence of electron acceptors used for benthic metabolism (O2, NO3-, Mn oxides, Fe oxides, SO42-) is evidently very close to the sediment-water interface. The oxic layer is less than 3 mm thick (3mm is our sampling resolution) in late summer and only reaches about 6 mm in winter, Fe is reduced at a depth of less than 3 mm in late summer, and H2S is abundant in pore waters beginning at a depth of 6-8 cm throughout the year. This is a site that was heavily contaminated due to its proximity to the Nut Island sewage outfall that was in operation from the early 1900s until 1998. The concentrations of Ag, Cu, and Pb remain at potentially damaging levels, although they are decreasing.
Our second study site (30 m water depth) is approximately 2 km west of the Massachusetts Bay outfall for Boston's sewage effluent (Figure 7.1). The annually averaged organic matter oxidation rate at this site is about half that at the Boston Harbor site, 600 µmol/cm2/y. Oxygen penetration depths range from about 3.5 to 5mm (Section 8, this report): the maximum penetration depth is similar to that at the Boston Harbor site, but the minimum depth is significantly deeper, with the result that our pore water data always show that there is no dissolved Fe in the upper 3 mm of the sediment. In addition, there is no H2S in pore waters in the upper 30 cm of the sediment column. The differences between this site and the Boston Harbor site, coupled with the fact that heavy metal concentrations in the sediments are much lower here, result in contrasting metal cycling in the two locations.
A conceptual model of the cycling of Ag, Cu, and Pb in coastal sediments.
Our work has shown that two phenomena dominate the cycling of Ag, Cu, and Pb in coastal sediments. First, their cycles are closely related to that of Fe. They are removed from solution by sorption onto freshly precipitated Fe oxides in the oxic sediment layer, and they are released to solution when microbes reduce Fe oxides. Second, they form highly insoluble sulfides, so that their cycles are also closely related to the S cycle.
The key to metal cycling in coastal sediments is an active sediment cycling layer in the upper 10 - 20 cm of the sediment column (Figure 10.1). The chemical characteristics of this layer are set to a large extent by the rate of input of organic matter, from both marine production and anthropogenic activities. The processing of the organic matter results in a sequence of layers in the sediments: an oxic layer is underlain by a layer in which Fe reduction and sulfate reduction occur. Fe reduction releases Fe2+ to the pore waters, and sulfate reduction releases H2S. The two products combine to form highly insoluble FeS. If sulfate reduction is rapid enough, FeS formation will remove all the released Fe2+, and H2S will accumulate. Both solute diffusion and rapid mixing of solids by animals living in the sediments can return chemically reduced species to the oxic layer, where they may be re-oxidized, to pass through the oxidation / reduction cycle many times before being buried. The intensity of the oxidation / reduction cycle is determined by the thickness of the sediment cycling layer, the presence or absence of dissolved H2S, and the mixing rate. Solute exchange across the sediment-water interface is central to this sedimentary cycling, as it both adds reactants (O2 and SO42-) from the water column and releases products, which may include heavy metals, back to seawater. A second mechanism of exchange with the overlying seawater, which appears to be at least as important to heavy metal cycling as solute diffusion, is sediment resuspension, which our results show contributes both solid and dissolved metals to the water column. How this Fe / S cycle interacts with anthropogenic metals depends in part on how the metals are added to the system: by deposition from the water column and, in previously contaminated sites, by mixing of contaminants into the cycling layer from below.
Heavy metal cycling and benthic / water column exchange at the Massachusetts Bay site:
The Massachusetts Bay site is characterized by an oxic layer that is 3-5 mm thick, an Fe reduction layer that extends to ~ 5 cm below the interface, and an absence of H2S in pore waters.
Differences between the behavior of the Ag, Cu, and Pb in the oxic layer reflect their differing affinities for attachment to Fe oxides. Pb is most strongly removed, and its concentration in oxic pore water rarely rises above its concentration in bottom water (Figure 10.2). Ag is often present and Cu is generally present at higher concentrations in surface pore water than in bottom water. All 3 metals are clearly released in conjunction with the reduction of Fe oxides in the 1 - 3 cm depth interval. Once in solution, they diffuse both upwards, toward the sediment-water interface, where they are removed when Fe oxides precipitate; and downwards, where they are removed to the solid phase. At the Massachusetts Bay site, where sulfate reduction is slow enough that dissolved H2S is not observed, upward diffusion is favored, resulting in enrichment of the metals in the solid phase at the sediment surface (Figure 10.3). Thus, although our work and that of Bothner (Section 7) show that there has been no increase in metal loading since the new outfall opened in Massachusetts Bay in September, 2000, diagenetic processes lead to an enrichment of metal concentrations in surface, relative to deeper, sediments. Particles that are enriched in Ag, Cu, and Pb are eroded preferentially when bottom currents are sufficient to resuspend particles: the first particles eroded are enriched over surface sediments by up to 40x for Ag, 8x for Cu, and 4.5x for Pb. Thus, resuspension is an important source of particulate metals to the water column. In addition, the resuspended particles release Ag and Cu to solution; both solute diffusion and resuspension are sources of dissolved metals to the water column. We are currently quantifying the relative roles of these processes by examining bottom current records to determine the frequency and strength of resuspension events in the study area.
Heavy metal cycling and benthic / water column exchange at the Hingham Bay site:
We noted above the differences in the active sedimentary cycling layer between the Hingham and Massachusetts Bay sites. The oxic layer is significantly thinner at Hingham in the summer. The sulfate reduction rate is high enough so that all Fe2+ is removed by precipitation of FeS and elevated concentrations of H2S are present in pore waters within 6 cm of the sediment-water interface. In addition, solid phase profiles of Ag, Cu, and Pb (Figure 10.4) show that the primary source of the metals to the cycling layer is upward mixing from previously deposited, contaminated sediments. At this site, the cycling layer has distinct seasonal characteristics (Figure 10.5). During the winter, the cycling system is "charged" by the downward migration of the oxic layer and upward mixing of sulfides. These processes result in oxidation of Fe, Ag, Cu, and Pb sulfides. The released Fe then precipitates as Fe oxides, to which trace metals adsorb readily. Ag and Cu become enriched in pore waters at the sediment-water interface in winter, but the less soluble Pb does not. The source of the enrichment may be the oxidation of sedimentary sulfides; another possible source is the decomposition of particles that have been deposited recently from the water column. During early summer, the trace metals are released to pore waters when Fe oxides are reduced. These metals can diffuse both upward and downward, but downward transport is favored because of the rapid formation of highly insoluble sulfide minerals. Thus, as the summer progresses, the recycled Fe oxides release less sorbed Ag, Cu, and Pb when they are reduced. These metals are no longer enriched in surface pore waters, and there is no longer a correlation between Fe2+ and the metals in pore waters. Winter and early summer conditions are compared in Figure 10.5. The largest fluxes of dissolved Cu and Ag from sediments to the water column occur in winter, during the "charging" phase of the seasonal cycle.
There is also important sediment / water column exchange of metals related to sediment resuspension. Our controlled erosion experiments, using an experimental chamber of the EROMES design (Tolhurst, 2000) show that (1) erosion results in significant release of particulate metals to the water column (Figure 10.6A), (2) short-term reactions lead to release of dissolved metals from the eroded particles (Figure 10.6B), and (3) the fine-grained particles that may remain in suspension for hours to days continue to release up to 6% of their metal loading to the water column over a 90 hour period (Figure 10.6C).
Benthic exchange and the budget of Cu in Boston Harbor:
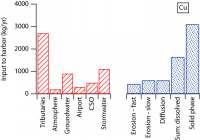
L. Kalnejais (WHOI) and R. Signell (USGS) used wind and tidal records to show that bottom currents at the Boston Harbor site were sufficient to erode sediments on the order of every 2 weeks, and that storm events could cause large erosion events on an annual basis. With this information, Kalnejais (2005) was able to show that sedimentary processes are a major part of the Cu cycle in Boston Harbor (Figure 10.7). The erosion tests indicated that release of particulate Pb is important, but dissolved Pb release was not. No data on inputs of Ag to the harbor are currently available.
The release of dissolved Cu from sediments (by erosion + diffusive flux) to harbor waters was shown to be larger than any other individual source except rivers (note that the river flux includes both dissolved and particulate Cu). The input of dissolved Cu from sediments was 60% of the input from rivers, and 30% of the sum of all other inputs. The total, annual input from sediments to harbor waters is ~ 80% of that from all other sources. It is important to note that the flux of solid Cu from sediments to harbor waters via erosion includes particles that are repeatedly resuspended and redeposited, so that the ultimate significance of this source is difficult to establish. However, a fraction of the eroded particles may remain in the water column for a long enough time for significant dispersion to occur. Eight hours after the completion of erosion experiments, 9% of the eroded mass, including 19% of the total particulate Ag eroded, 15% of Cu, and 34% of Pb, remained in suspension.
The importance of the sedimentary source will increase as pollution control reduces the other sources. The legacy of contaminated sediments from earlier activities will be increasingly important to the cycles of contaminants in Boston Harbor.
|
Click on figures below for larger images (PDF format).
 | Figure 10.1.
|
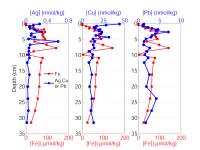 | Figure 10.2.
|
 | Figure 10.3.
|
 | Figure 10.4.
|
 | Figure 10.5.
|
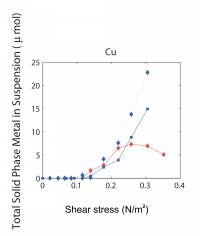 | Figure 10.6A.
|
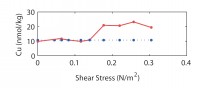 | Figure 10.6B.
|
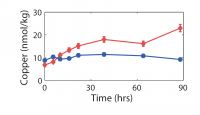 | Figure 10.6C.
|
 | Figure 10.7.
|
|
